Introduction
Streptomyces coelicolor A3(2) is one of the best studied representatives amongst other members of the genus Streptomyces [1]. Like the streptomyces genus in general, it lives in soil [2]. It is considered a model organism to study soil bacteria [3], which has been studied genetically for about 60 years [4]. They have the capability to degrade chitin and other compounds that are difficult to degrade which makes them especially important [5]. This bacterium produces a range of secondary metabolites, including actinorhodin, undecylprodigiosin, calcium-dependent antibiotic, methylenomycin A and perimycin [6]. Some of them have antifungal activities also. So, Streptomyces coelicolor A3(2) has the potential to make such secondary metabolites, and metagenomic analysis has revealed it has tremendous quantities of significant biosynthetic gene sets [7,8].
These characteristics have elicited biotechnological interest in this bacterium and have aroused the interest of researchers in the past few years to investigate the different proteins involved in secondary metabolites production. As an example, it is recently found that albaflavenone, germicidin A, and chalcone are produced during germination of Streptomyces coelicolor [9] and the genes responsible for the biosynthesis of streptomycete secondary metabolites are generally clustered with high expression of regulation [10]. Another research shows that a group of mtbH-like genes in S. coelicolor are necessary for some secondary metabolite production [11]. Streptomyces coelicolor has three such genes, cloY is one of them [11]. When all three genes were absent, clorobiocin, an antibiotic which inhibits the enzyme DNA gyrase was produced only in very small amounts, but when cloY was restored, clorobiocin was produced at a more significant level [11].
Streptomyces coelicolor A3(2) is reported to have 8,667,507 base pair linear chromosome, containing the largest number of genes so far discovered in a bacterium [10]. The genes so far predicted are 7,825 which include more than 20 clusters coding for known or predicted secondary metabolites [10]. However, there are many proteins of this bacterium which are considered hypothetical proteins as their structures and biological functions are not yet known. These proteins can be very important and their annotation can lead to knowledge about new structures, pathways, and functions. Thus, bioinformatics approaches can play an important role in predicting and analyzing various forms of structure of those hypothetical proteins, their biological functions as well as protein-protein interactions.
With the advancement of in-silico analysis, it became easier to annotate function to a hypothetical protein using various bioinformatic tools. Thus, the purpose of this study was to assign structural and biological function to the hypothetical protein SCO0618 (accession No. NP_624929.1) of S. coelicolor for an improved understanding of the protein. Subcellular localization, secondary structure, and active site were predicted and protein-protein interaction was analyzed. Further, a good quality model of the SCO0618 was tried to generate using homology modeling techniques.
Methods
Sequence retrieval and similarity identification
The sequence information of the hypothetical protein (NP_624929.1) was retrieved from the NCBI database. The sequence was then collected as a FASTA format sequence and submitted to several prediction servers for the in-silico characterization (Table 1). To get the initial prediction about the function of the targeted hypothetical protein, similarity search was performed with the NCBI protein Database (https://www.ncbi.nlm.nih.gov/) against nonredundant and SwissProt [12] database to find out the proteins that might have structural similarities with that of the uncharacterized protein by using BLASTp program [13].
Multiple sequence alignment and phylogeny analysis
Multiple sequence alignment was performed using MUSCLE server of EBI (https://www.ebi.ac.uk/Tools/msa/muscle/) [14] and visualized using the CLC Sequence Viewer 7.0.2 (http://www.clcbio.com). The phylogeny analysis was done by using the webtool Phylogeny.fr (http://phylogeny.lirmm.fr/) [15].
Physiochemical properties analysis
The physical and chemical properties including molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, total number of negatively charged residues (Asp + Glu), total number of positively charged residues (Arg + Lys), instability index, aliphatic index, and grand average of hydropathicity (GRAVY) predictions, etc. were performed by the ProtParam (http://web.expasy.org/protparam/) [16] tool of ExPASy.
Conserved domain, motif, fold, coil, family, and superfamily identification
Search carried out at conserved domain database (CDD, available at NCBI) [24], for conserved domain. Protein motif search was carried out using Motif (Genome Net) server [25]. Pfam [26] and SuperFamily [27] database searches were done to assign the protein’s evolutionary relationships. For the detection of coiled-coil conformation within the protein, the COILS server [28] was employed. Protein sequence analysis and classification server InterProScan [29] was employed for the functional analysis of the protein. For protein folding pattern recognition, PFP-FunD SeqE server [30] was used. And STRING 10.0 [31] search was carried out for the identification of possible functional interaction network of the protein.
Three-dimensional structure prediction
The three-dimensional structure was predicted by HHpred server (https://toolkit.tuebingen.mpg.de/tools/hhpred) [34] of the Max Planck Institute for Developmental Biology, Tübingen which is based on the pairwise comparison profile of hidden Markov models (HMMs). For higher accuracy, the 3D structure was predicted on the basis of best scoring template. Later the 3D structure was refined through YASARA energy minimization server [35].
Model quality assessment
Finally, PROCHECK (https://servicesn.mbi.ucla.edu/PROCHECK/) [36], Verify3D (http://nihserver.mbi.ucla.edu/Verify_3D/) [37], and ERRAT Structure Evaluation server (https://servicesn.mbi.ucla.edu/ERRAT/) [38] were used for quality assessment of the predicted three dimensional structure.
Active site detection
The active site of the protein was determined by the Computed Atlas of Surface Topography of Protein (CASTp) (http://sts.bioengr.uic.edu/castp/) [39] which provides an online resource for locating, delineating, and measuring concave surface regions on three-dimensional structures of proteins.
Results and Discussion
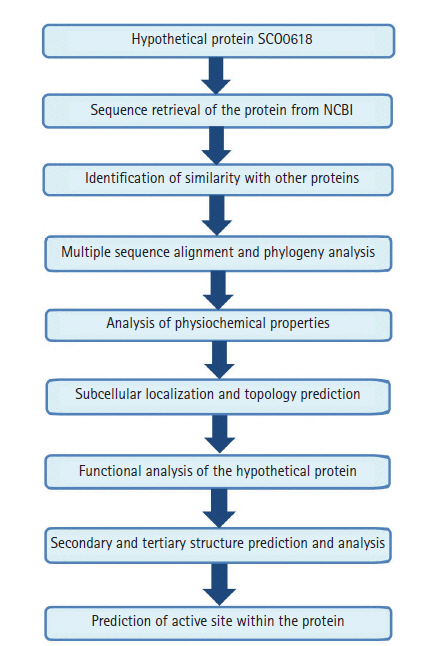
The work-flow of the study was shown in Fig. 1.
Sequence and similarity information
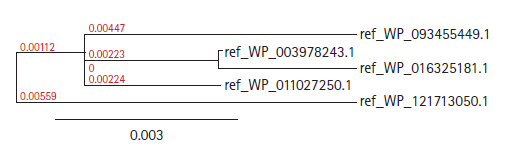
The Blastp result against non-redundant and SwissProt database showed homology with other hydrolase and sulfurtransferase proteins (Tables 2 and 3). Multiple sequence alignment (Supplementary Fig. 1) was considered the FASTA sequences of the hypothetical protein (SCO0618) and the homologous annotated proteins. For the confirmation of homology assessment between the proteins, down to the complex and subunit level, phylogenetic analysis was also performed. Phylogenetic tree was constructed based on the alignment and BLAST result which gives the similar concept about the protein (Fig. 2). The distances between branches are also included.
Physicochemical features
The protein consist of 461 amino acids, among the most abundant were Ala (92) followed by Val (51), Arg (42), Gly (41), Leu (40), Asp (32), Glu (30), Pro (26), Thr (21), Ser (19), His (17), Phe (11), Ile (10), Tyr (8), Trp (6), Asn (5), Gln (4), Met (4), and Cys (2). The calculated molecular weight was 48216.15 Da and theoretical pI was 5.27 indicating the protein to be negatively charged. Total number of positively charged residues (Arg + Lys) and the total number of negatively charged residues (Asp + Glu) were found to be 62 and 42, respectively. The computed instability index was 32.67 classifying the protein as stable one. Aliphatic index was 94.34 which gives an indication of proteins’ stability over a wide temperature range. The grand average of hydropathicity (GRAVY) was 0.053. Positive value of GRAVY indicates that the protein is polar. Protein half-life computed was found to be 30 h in mammalian reticulocytes (in vitro), >20 hours in yeast (in vivo), >10 h in Escherichia coli (in vivo). And the molecular formula of protein was identified as C2119H3350N636O643S6.
Functional annotation of the hypothetical protein
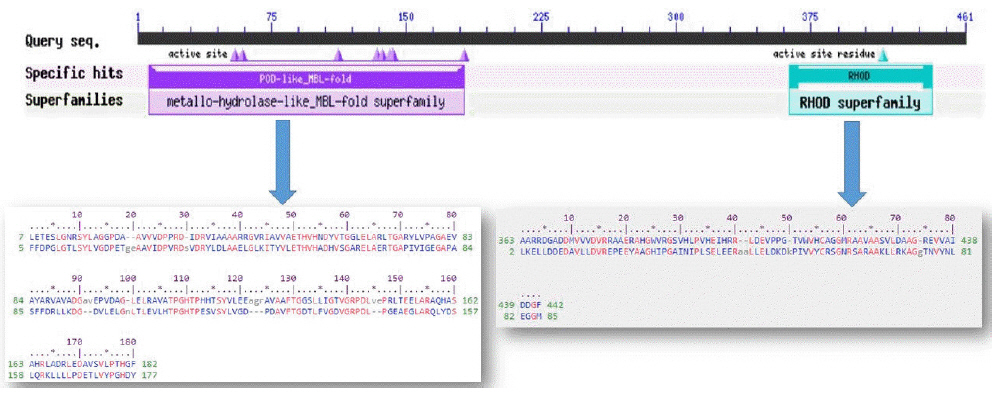
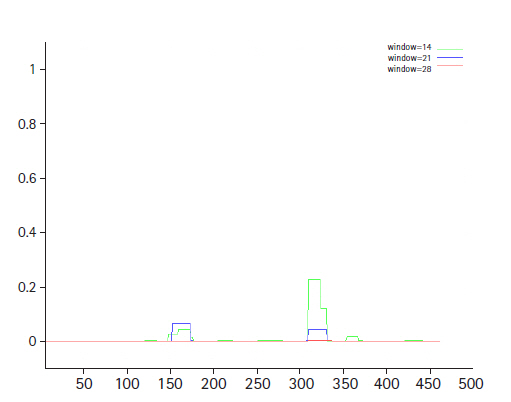
The conserved domain search tool revealed that this hypothetical protein sequence was found to have two domains, MBL-fold metallo-hydrolase domain (accession No. cd07724) and rhodanese homology domain (RHOD) (accession No. cd00158). The result was also checked by two other domain searching tools namely InterProScan and Pfam. Pfam server predicted the Rhodanese like domain at 362–444 amino acid residues with an e-value of 2.3e-05 and Metallo-beta-lactamase superfamily domain at 16–171 amino acid residues with an e-value of 4.7e-07. InterproScan server predicted Rhodanese like domain at 249–454 amino acid residues and Metallo-beta-lactamas domain at 13–180 amino acid residues. Rhodanese like domain, lactamase-B and MreB-Mbl domains were also found by Motif server. Superfamily search revealed present of Metallo-hydrolase/oxidoreductase and rhodanese/cell cycle control phosphatase superfamily. β-Lactamases can catalyze the hydrolysis of a wide range of β-lactam antibiotics. Members of the MBL-fold metallohydrolase superfamily are mainly hydrolytic enzymes which carry out various biological functions. Both the active and inactive version of the Rhodanese domain in a variety of proteins including certain protein phosphatases, sulfide dehydrogenases, certain stress proteins and sulfuryl transferases, where they are thought to play a regulatory role in multidomain proteins (Fig. 3). All these results confirm the presence of hydrolytic enzyme containing domains in this protein. Fold pattern recognition by PFP-FunDSeqE tool revealed the presence of a ‘(TIM)-barrel’ fold within the protein sequence. (TIM)-barrel structure is generally eight stranded α/β barrel. The x-axis of the graph represents the position in the protein of amino acid number (starting at the N-terminus) and the y-axis shows the coiled coil whereas ‘Window’ refers to the width of the amino acid ‘window’ that is scanned at one time (Fig. 4).
Subcellular localization nature
Subcellular localization analysis was predicted by CELLO and validated by PSORTb, SOSUIGramN, and PSLpred. The subcellular localization of the hypothetical protein was predicted to be a cytoplasmic protein (Table 4). Absent of transmembrane helices predicted by THMM and HMMTOP also emphasizes the result of being a cytoplasmic protein. Also, CCTOP server predicted that the query protein was not a transmembrane protein. All these results summarize the protein as a cytoplasmic one.
Secondary structure analysis
The SOPMA secondary structure prediction server analysis revealed the proportions of alpha helix, beta turn, extended strand, and the random coil of protein as 31.89%, 9.11%, 18.87%, and 40.13%, respectively (Supplementary Fig. 2).
Three-dimensional structure analysis
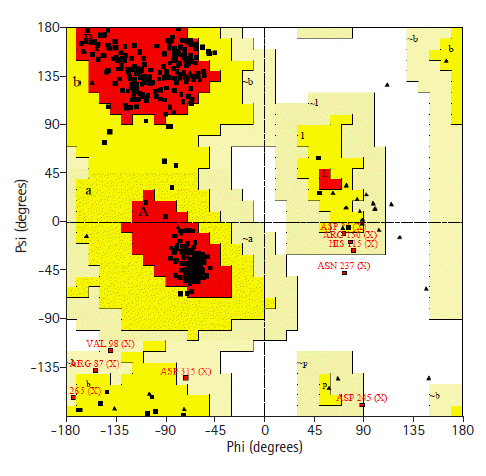
Prediction of 3D structure was done by HHPRED server. The server predicted 3D structure of the protein with 100% identity with the highest scoring template (PDB ID: 3TP9_A) (Fig. 5). 3TP9 is the crystal structure of Alicyclobacillus acidocaldarius protein with β-lactamase and rhodanese domains. This protein is a homo-dimer which has two chains (chain A and chain B) and the chain A was used as template to build the model. Validation of the predicted three-dimensional model was assessed by PROCHECK through Ramachandran plot analysis, where the distribution of φ and ψ angle in the model within the limits are shown (Table 5, Fig. 6). Residues in the most favored regions covered 90.9%, which is the quality of a valid model. Finally, the established model of 3D structure for the target sequence was verified by structure validation server Verifiy3D and ERRAT. In the Verify3D graph, 92.73% of the residues have averaged 3D-1D score ≥ 0.2 which indicates that the environmental profile of the model is good and the overall quality factor predicted by the ERRAT server was 69.0583 indicates a good model. The 3D structure was later modified by YASARA energy minimization server. The energy calculated before energy minimization was –77,930.2 kJ/mol whereas after energy minimization (through 3 round of steepest descent method), it was changed to far less value of –244,148.6 kJ/Mol making the modeled structure more stable one.
Protein-protein interaction analysis
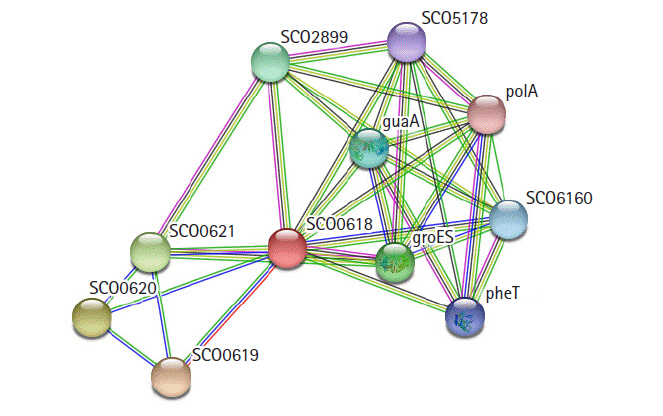
STRING 10.0 search was carried out for the identification of possible functional interaction network of the protein [31]. The identified functional partners with scores were; SCO0619 (0.970), SCO0620 (0.743), SCO0621 (0.739), groES (0.568), SCO2899 (0.568), guaA (0.545), SCO6160 (0.520), pheT (0.508), SCO5178 (0.485), polA (0.473). Of them, SCO0619 is a possible membrane protein. The others are two hypothetical proteins, two chaperonins, GMP synthase, multifunctional fusion protein, phenylalanine tRNA ligase β subunit, putative sulfurylase, and DNA polymerase I (Fig. 7).
Active site of the hypothetical protein
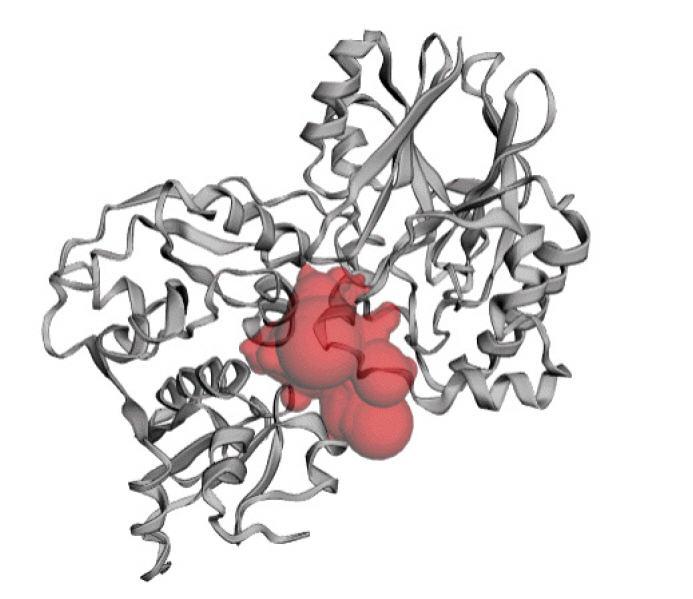
The predicted active site of the protein found that 42 amino acids are involved in potent active site (Fig. 8). The best active site was found in areas with 613.075 and a volume of 608.774 amino acids. The amino acid residues in the active site were shown in Supplementary Fig. 3.
Conclusion
The identification of protein functions is fundamental for the understanding of biological processes. So, this study was aimed to determine the structural and biological function of SCO0618, a hypothetical protein of this bacterium through an in-silico approach. The identified protein revealed several characteristics such as cytoplasmic nature, hydrolytic enzymes containing domain presence, ‘(TIM)-barrel’ fold presence, and hydrolase activity emphasize the significance of this protein. These characters of the hypothetical protein will strengthen basic knowledge on S. coelicolor. So, extended in-vitro research has to be carried out to experimentally validate the possibilities shown here and to find out the proteins’ role in biotechnology.