Availability: HisCoM-PCA is available on the website (http://statgen.snu.ac.kr/software/HisCom-PCA/index.html).
Introduction
In genome-wide association studies (GWAS), researchers have identified many single-nucleotide polymorphisms (SNPs) associated with the traits of interest (phenotypes) [1]. However, these SNPs sometimes may sometimes suffer from a lack of biological interpretation [2]. To enhance interpretation of SNP association results, many gene-based analysis and pathway-based analysis have been widely performed in GWAS. For examples, PHARAOH was developed for pathway analysis of rare variants, and hierarchical structural component analysis of gene-gene interactions (HisCoM-GGI) was proposed for gene-gene interaction analysis of common variants [3,4].
Recently, we presented a hierarchical structural component model (HisCoM) for pathway analysis of common variants (HisCoM-PCA) to identify pathways associated with traits [5]. HisCoM-PCA is based on principal component analysis (PCA) for dimension reduction of SNPs in each gene, and the HisCoM for pathway analysis. In the dimensional reduction step of HisCoM-PCA, various principle component scores (PC) selection criteria may be used. The criterion may be defined by a threshold of cumulative proportion of variances. It can also be defined by using only the first PC for each gene. In the pathway analysis step, multiple published pathway databases and specific combination of pathways can be used to identify pathways associated with the traits of interest.
In our previous study, we used only pathway information of the Kyoto Encyclopedia of Genes and Genome pathway database [6] and adopted two criteria to select PC: (1) using only the first PC and (2) using the PCs whose cumulative proportion of variances are more than 30%. To enable researchers to perform various pathway analysis, we developed HisCoM-PCA software for allowing the users to set the PC selection criterion and pathway information flexibly.
Implementation
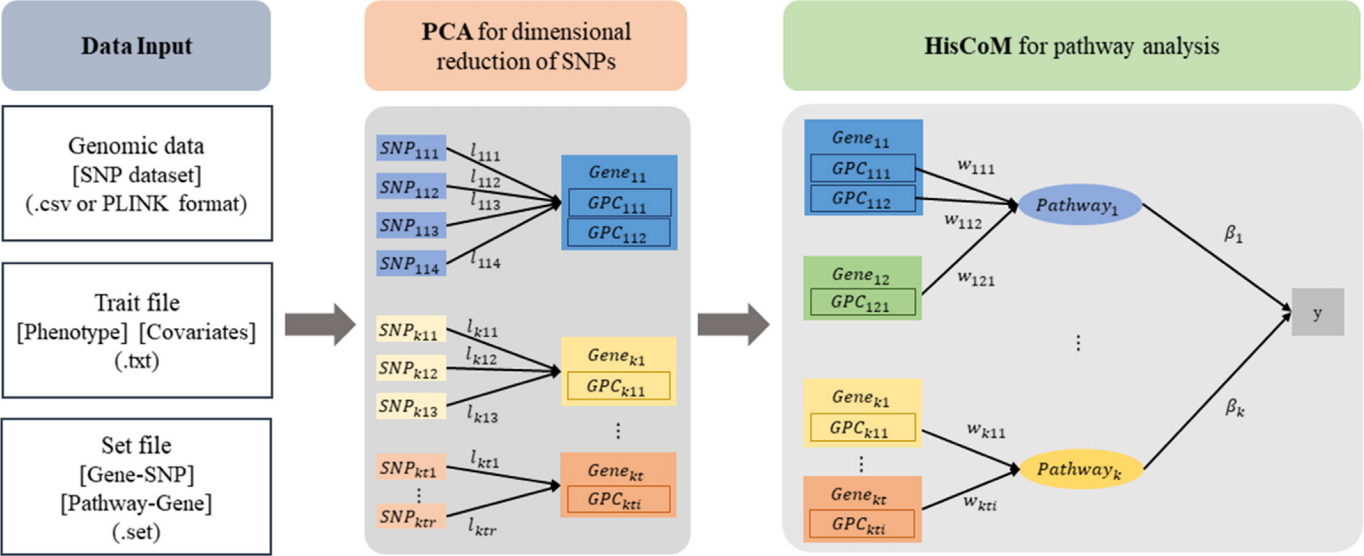
The workflow of the HisCoM-PCA software is shown in Fig. 1. The HisCoM-PCA mehod has been proposed for pathway analysis of common variants by constructing a hierarchical model using SNP-gene-pathway information. The HisCoM-PCA method consists of two steps: (1) dimensional reduction of SNPs by PCA and (2) pathway analysis with a hierarchical component model. In the first dimensional reduction step of HisCoM-PCA software, the user can define the number of PCs for each gene by one of the following two options: (1) the threshold of cumulative proportion of variances and (2) only the first PC. Using the selected PCs, pathway analysis can be performed based on the user-entered pathway datasets. In the second pathway analysis step, HisCoM-PCA utilizes ridge-type penalization and performs a permutation test to estimate the gene and pathway effects on the phenotypes.
Input file
The HisCoM-PCA software takes four inputs: (1) a SNP dataset in csv file or PLINK format files, (2) a txt file with phenotype and covariate(s), (3) a set file that consists of two columns for gene name and SNP id, and (4) a set file that consists of two columns for pathway name and gene name. Furthermore, the program also accepts the published pathway databases in MsigDB [7,8].
Output file
The HisCoM-PCA program generates the following output files: (1) a ŌĆś[prefix]. gene-pca_summary.csvŌĆÖ file that contains the number of SNPs and PCs for each gene, (2) a ŌĆś[prefix].gene.ressum.csvŌĆÖ file that contains pathway name, gene name, number of permutation, weight of gene for the pathway, gene coefficient, and permutation p-value of gene, and (3) a ŌĆÖ[prefix].pathway.ressum.csvŌĆÖ file that contains pathway name, number of permutation, number of genes in each pathway, pathway coefficient, and permutation p-value of pathway.
Conclusion
We introduced our HisCoM-PCA software for pathway analysis of common variants in GWAS. HisCoM-PCA software supports pathway analyses using multiple candidate pathways and customized PC selection criterion. We expect that our HisCoM-PCA software is useful for users to perform various pathway analyses. This section should contain sufficient detail so that all procedures can be repeated, in conjunction with the cited references. The manufacturer and model number should be stated in this sectionŌĆöfor example, as Sigma Chemical Co. (St. Louis, MO, USA).