Availability: HisCoM-mimi is available at http://statgen.snu.ac.kr/software/hiscom-mimi/.
Introduction
miRNA is a well-known form of noncoding RNA that affects biological mechanisms by regulating the expression of target mRNA. Many researchers have found that cancer cells and normal cells exhibit different inhibition mechanisms, suggesting that miRNAs could be used as biological markers for the diagnosis of cancer [1-3]. In our previous study, we presented a hierarchical structural component model (HisCoM-mimi) to find the miRNA-mRNA interaction pairs associated with binary phenotypes that could be candidates for cancer diagnosis biomarkers with an interpretable biological inhibition mechanism. Recently, many findings regarding the target mRNAs of miRNAs have been incorporated into various databases. TargetScan is one such database with recently updated findings [4]. The basic principle used by TargetScan to predict the mRNAs that miRNAs target for inhibition is to compare the sequences of untranslated mRNA regions to those of miRNAs [5]. However, many studies have shown that miRNAs select their target mRNAs based not only on the similarity of sequences, but also on other structural findings [6]. Thus, miRNA-mRNA integration analysis requires experimental confirmation of which mRNAs are truly inactivated by miRNAs. MiRTarBase is a database that collects experimental findings [6]. In our previous study, we only used TargetScan to find pairs of miRNA-mRNA relationships [7]. To enable researchers to utilize more flexibly information regarding the target mRNAs inhibited by miRNAs, we added miRTarBase database information to our software.
Implementation
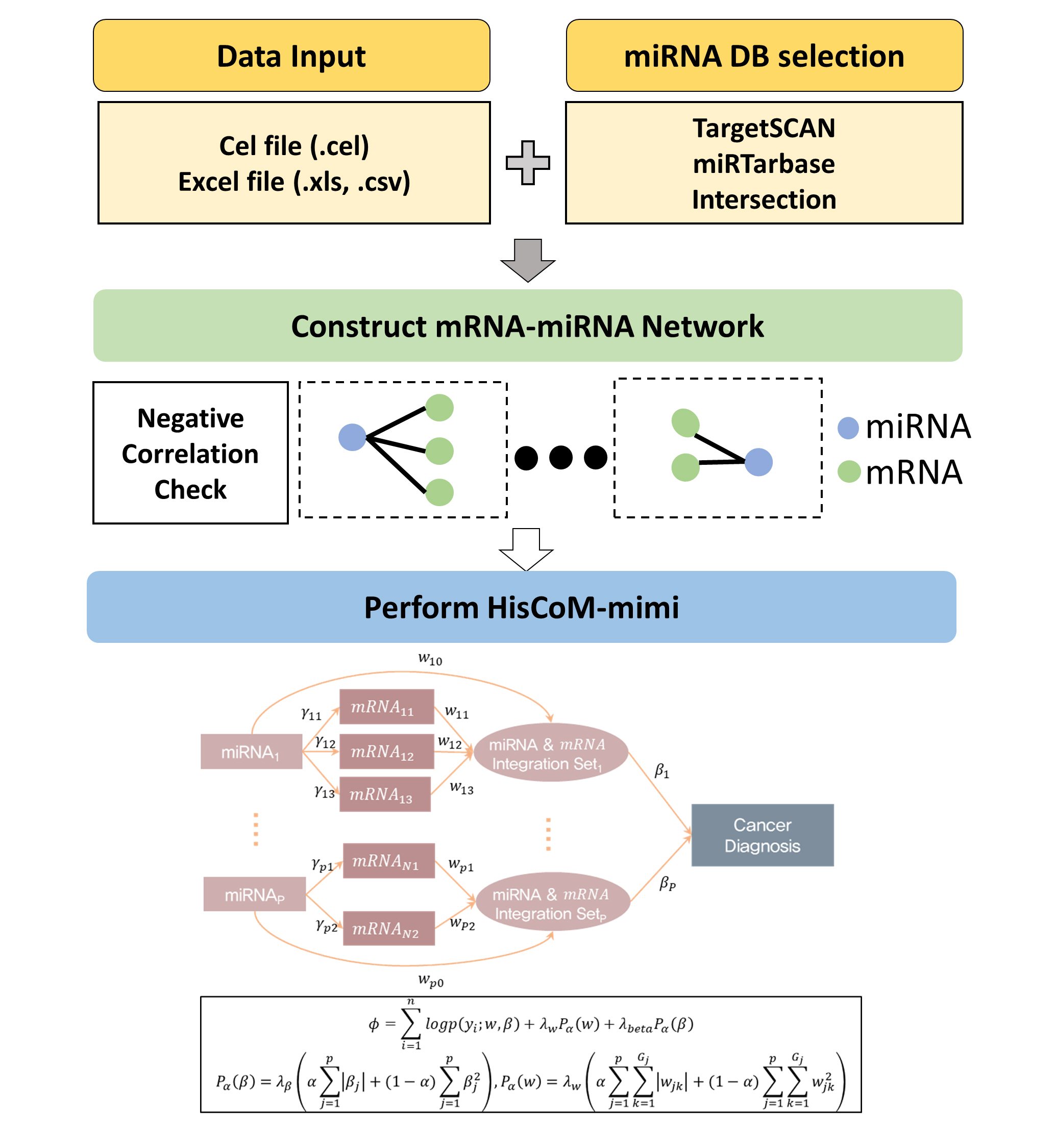
Fig. 1 shows the hierarchical structural component analysis workflow for the HisCoM-mimi application, which requires miRNA and mRNA expression datasets and additional files (phenotype and covariates). The program now accepts two formats (miRNA and mRNA CEL files or an Excel-type expression dataset).
Next, miRNA-mRNA networks are constructed by combining the miRNA database information and correlation coefficients computed based on the user-entered datasets. Users can select an miRNA database in three ways: TargetScan results, miRTarBase results, and the intersection of both databases. The user can define the filtering network criteria by two options: (1) the choice of the databases and (2) a p-value threshold for the correlation coefficients between miRNAs and mRNAs.
After constructing an mRNA-miRNA integration set, HisCoM-mimi can be performed. HisCoM-mimi can accommodate not only the ridge penalty, but also the lasso and elastic-net penalties, which result in reduced computing time and a smaller number of nonzero coefficients [8, 9]. A cross-validation procedure is necessary to find the optimal penalties that maximize the log likelihood of the validation set [7].