Introduction
In the past decade, genome-wide association studies (GWASs) have successfully identified genetic variants associated with human traits and complex diseases. However, traditional GWASs have several issues and limitations that do not completely explain the heritability of complex human diseases or related traitsŌĆöso-called missing heritability [1]. To address these issues, various approaches have been proposed, such as rare variant association analysis, genetic interactions, and epistasis [1]. Among possible explanations for missing heritability, there has been growing consideration of gene-gene interaction (GGI) analysis for common genetic variants in terms of statistical approaches and analysis software. Although most research models focus on the case-control study design, there are not many software programs that can analyze a continuous phenotype. Furthermore, GGI analysis has several advantages in terms of statistical power, computing performance, and biological interpretation over single nucleotide polymorphism (SNP)ŌĆÆSNP interaction (SSI) analysis, but there is not much analysis software to analyze it.
In this study, we developed software for ŌĆ£Hierarchical Structural Component Analysis of Gene-Gene InteractionsŌĆØ (HisCoM-GGI) [2] that can analyze both GGI and SSI analyses for a continuous phenotype. The basic framework of the HisCoM-GGI software was ŌĆ£Workbench for Integrated Superfast Association study with Related DataŌĆØ (WISARD) [3], which accepts various types of input formats and provides quality control or data management options that can be used to filter out individuals or genetic variants. In addition, the WISARD program supports multithreading to improve the efficiency of the analysis. By using the advantages of WISARD, HisCoM-GGI software includes all of the functions for GGI analysis on a genomewide scale, such as input format support, data preprocessing, and multithreading.
Implementation
The workflow of the HisCoM-GGI software is shown in Fig. 1. The HisCoM-GGI method is based on the Generalized Structured Component Analysis (GSCA) method [4]. GSCA is a component-based approach to structural equation modeling. The latent variables of GSCA are defined as components and are estimated by the weighted sums of the observed variables. The HisCoM-GGI method has been proposed for genetic interaction analysis by constructing a hierarchical model that consists of SNP-level and gene-level summaries. Therefore, the HisCoM-GGI method provides the results of SSI and GGI in a single model. The HisCoM-GGI software was implemented in C++ and was developed using programs for Linux and Windows.
Input file
The HisCoM-GGI software takes three inputs: (1) a genomic dataset in PLINK or variant calling format format, (2) trait information on the covariates and phenotypes, and (3) a set file that consists of two columns for gene name and SNP ID (rsID), respectively. Furthermore, users can optionally specify a gene-gene pair list to analyze.
Output file
The HisCoM-GGI program generates the following files: (1) a ŌĆś[prefix].gesca.latent.resŌĆÖ file that contains p-values of gene-level interactions and (2) a ŌĆś[prefix].gesca.manifest. resŌĆÖ file that contains p-values of SNP-level interactions.
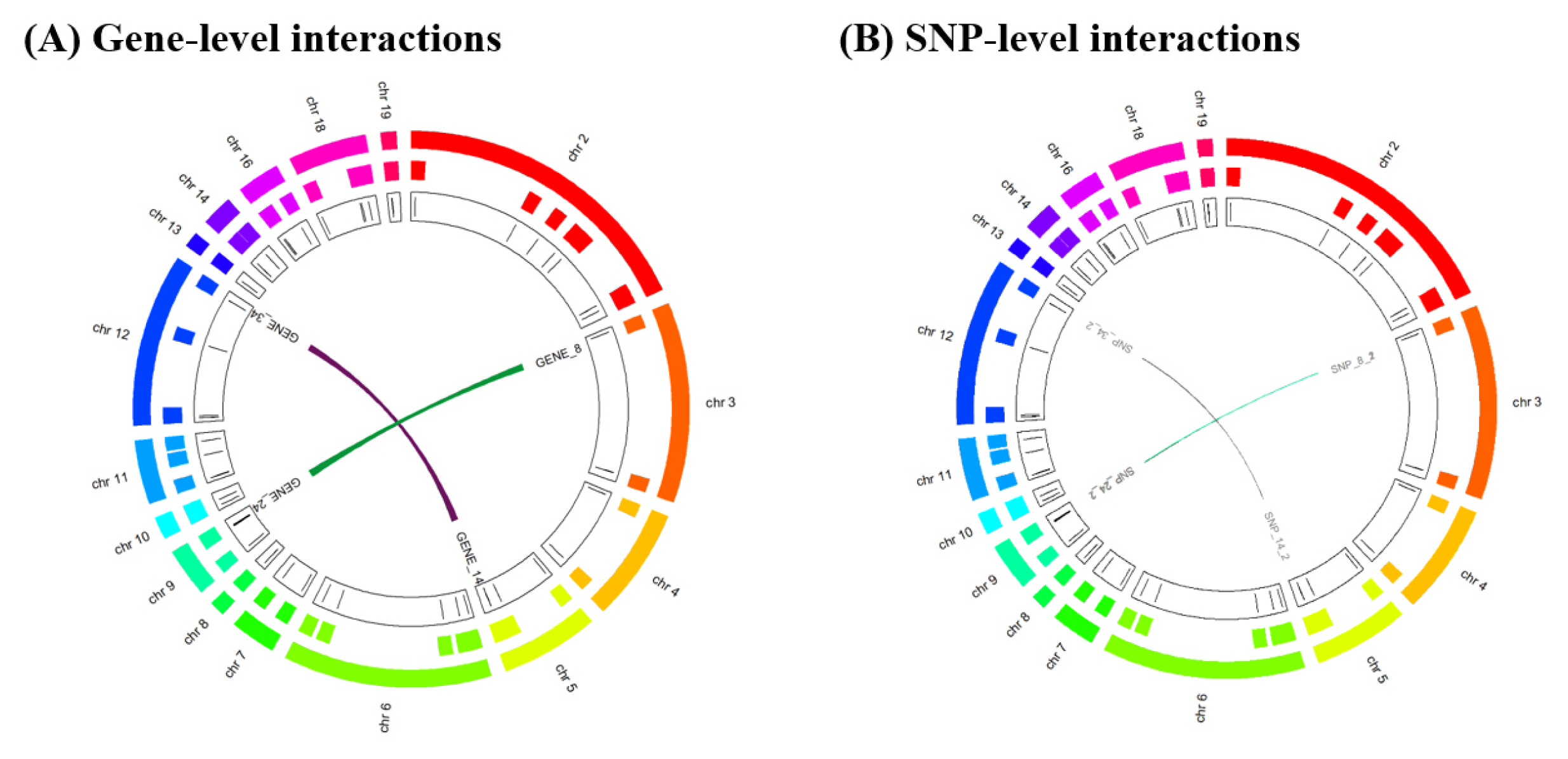
Visualization of results
We provide the circos plots for the results of GGI and SSI using R script at the following website: http://statgen.snu.ac.kr/software/hiscom-ggi/?page_id=10. The visualized results of the HisCoM-GGI software are shown in Fig. 2.
Conclusion
In this paper, we introduce the HisCoM-GGI software program, which provides a statistical test of identifying gene-level and SNP-level interactions for continuous phenotypes. We conclude that HisCoM-GGI software may be a valuable tool for the identification of genetic interactions, allowing us to better understand biological mechanisms of complex human diseases or related traits. The software is freely available with a tutorial dataset through the website http://statgen.snu.ac.kr/software/hiscom-ggi.