Introduction
High-throughput mRNA sequencing technology has developed at great pace in recent years [1]. Data from RNA sequencing (RNA-Seq) experiments across many species and tissue types are available for free access through public repositories. While RNA-Seq data have a wide range of applications, such as alternative splicing research, fusion gene finding, novel transcript discovery, etc., the most important and widely considered application is the quantification of gene expression profiles and the assessment of differentially expressed genes (DEGs) [2].
Evaluating differential expression in conditions by RNA-Seq is a multi-step process [3]. R/bioconductor [4] has been used to develop tools for the statistical analysis of RNA-Seq data. Some packages stem from classical methods for microarray data analysis, like the t test. Others, like edgeR [5], DESeq [6], DEGSeq [7], and baySeq [8], have recently been developed to the characteristics of RNA-Seq data. However, different packages partially support varying steps of the multi-step process in a very inconsistent manner. Moreover, no R packages support data filtering steps to improve the statistical power and control outliers that might have an undesirable influence on further analysis [9].
This study proposes TRAPR (Total RNA-Seq Analysis Package for R, http://www.snubi.org/software/trapr), an integrated pipeline for the analysis of RNA-Seq gene expression data. TRAPR uses gene expression tables to perform all RNA-Seq analyses, including data preprocessing, filtering, normalization, and statistical tests. TRAPR also provides visualization functions for data exploration and results' summarization. TRAPR provides a unique way of combining state-of-the-art analysis methods in an integrated pipeline for comprehensive RNA-Seq data analysis. For instance, upper-quartile normalization followed by zero-value filtering, variance stabilizing normalization (VSN) [10], and edgeR statistical testing with proper data visualization can easily be streamlined. These combinations have considerable potential to improve the accuracy and statistical power [11] of the analysis of RNA-Seq gene expression data.
Results
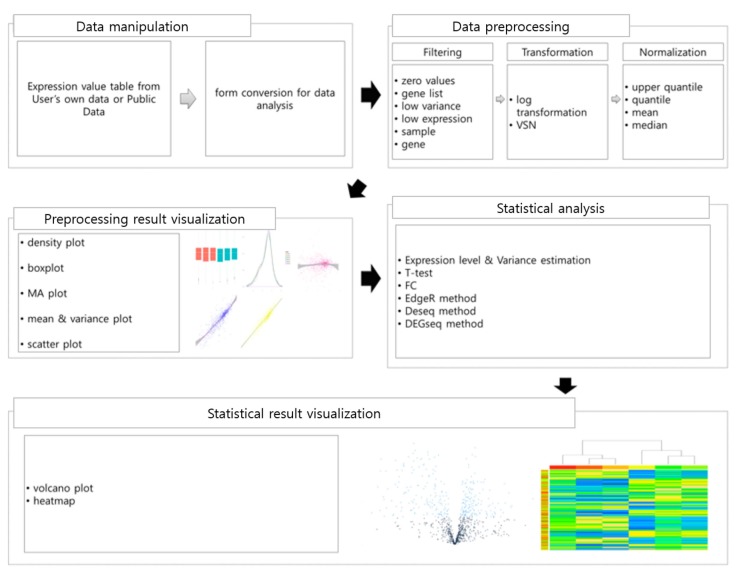
Fig. 1 shows the five steps of TRAPR, i.e., data manipulation, data preprocessing, statistical analysis, preprocessing result visualization, and statistical result visualization.
Data manipulation
TRAPR provides two functions to import RNA-Seq experimental data and four functions to export results to files. TRAPR can read text files for expression data as well as for a list of genes. During or following analysis, users can export DEG lists or detailed tables for DEG and expression tables, which other tools can utilize.
Data preprocessing
TRAPR provides three types of data preprocessing methods: filtering, transformation, and normalization. TRAPR filtering has six filter types: sample, gene, zero value, low variance, low expression, and gene list. Unlike DNA microarrays that have a fixed number of probes, RNA-Seq explores massive amounts of isoforms and novel transcripts mixed with noise, such that it returns many zeros and nonsense values. Genes encoding miRNAs or snoRNAs often show extremely high expression levels, even though they are treated by a poly-A purification procedure. These outliers can easily be removed by zero-value and gene filters. Statistical power can be improved by low-expression and low-variance filters by reducing non-standard distributions. Analyzing different combinations of samples can conveniently be supported by sample filtering.
TRAPR provides two well-known transformation functions, log2 transformation and VSN, followed by hyperbolic arcsin, arcsin(x), and transformation [11] to standardize data distribution and normalize variance distribution, respectively.
TRAPR provides many normalization methods, including upper quantile [12], quantile, mean, and median normalizations. One can conveniently compare the effect of applying different methods with the following statistical testing and visualization functions.
Statistical testing
TRAPR has several statistical testing functions to identify DEGs. Student t-test and statistical methods suggested in edgeR, baySeq, and DESeq assume a normal distribution or a Poisson distribution. Meanwhile, methods in DEGseq and NOISeq [12] do not need to make any assumption. For now, there is no correct answer that fits the characteristics of RNA-Seq data, and each method has its own merits. TRAPR allows users to choose their own methods to build customized analysis pipelines. The t test is recommended for large datasets, whereas edgeR and simple-fold change work for datasets with a small number of samples. DEGs can be labeled and saved as files containing lists of gene names with detailed information.
Visualization
Data preprocessing steps are not supported by visualization functions in previously developed packages, while proper visualization is essential and powerful for evaluating the quality of the RNA-Seq data and the preprocessing steps. TRAPR provides five flexible plotting functions, including density, boxplot, MA, scatter, and meanâvariance plots. Volcano plots and heatmaps are also provided to visualize the results of statistical analysis. Each visualization function has direct access to FPKM values and differential expression values.
Discussion
We have developed TRAPR, an R package for RNA-Seq data analysis. TRAPR provides an entire pipeline for RNA-Seq analysis, which is not merely a combination of currently available tools, but the backbone that facilitates the proper application and coordination of these tools. For instance, upper-quartile normalization followed by zero-value filtering, VSN, and edgeR statistical testing with proper data visualization can easily be streamlined through TRAPR. These combinations will help improve accuracy and statistical power. TRAPR provides visualization tools and file I/O functions to evaluate the quality and characteristics of the data. TRAPR was developed and integrated in R, such that it can be easily applied to other technologies like Serial Analysis of Gene Expression and microarray. Various filters have been integrated into the package. TRAPR can be used as a platform to interweave RNA-Seq data analysis tools and packages to take advantage of the virtues of each.