Introduction
Genomics and proteomics data are typically analyzed by hierarchical clustering, followed by visualization with heatmaps [1-3]. Various algorithms have been implemented in the data clustering procedure [4]. The visualization of clustered data includes tree-based hierarchical clustering patterns and heatmaps of experimental values [5]. Simultaneously carrying out clustering and visualization in a single platform provides a convenient tool for choosing an appropriate clustering algorithm and finding patterns in the resulting heatmaps. Previously, bioinformaticists used programmable tools, such as R and Matlab, and commercial data-mining packages to analyze their data. A simple and integrated program will allow experimental scientists to intuitively identify meaningful patterns from a large dataset without requiring knowledge of scripting computer languages or statistical theory.
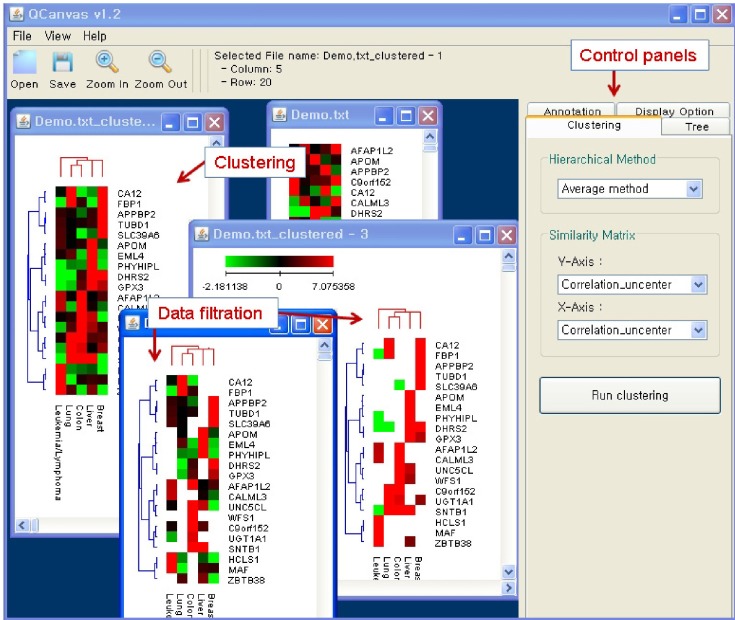
Herein, we introduce a user-friendly tool, QCanvas, which integrates diverse clustering algorithms and an interactive heatmap display interface (Fig. 1). This program directly imports raw experimental data in a matrix format and displays these data in a heatmap. Various clustering methods can be applied to two-dimensional data, with the real-time generation of clustered heatmaps. Furthermore, subsets of heatmap data can be selectively displayed, based on user-defined filters. QCanvas is an easy-to-use and powerful tool for fast data analysis and interpretation by bench scientists. Without any knowledge of scripting languages and without any graphics-editing software, one can generate and customize tree-clustered heatmaps with high-quality graphics.
QCanvas: Implementation and Functions
Data clustering
QCanvas provides a total of eight popular measures for generating the similarity matrix-i.e., Correlation uncenter, Correlation center, Absolute corr-uncenter, Absolute corrcenter, Spearman rank, Kendall's tau, Euclidean distance, and City-block distance. All of these measures have typically been included among the data clustering methods of previous tools [4]. In QCanvas, the calculation of the similarity matrix is selectively applied to the data for the x-axis and the y-axis independently. Hierarchical clustering is simultaneously carried out based on the established similarity matrices. QCanvas provides diverse algorithms for hierarchical clustering, such as the average method, centroid method, single method, and complete method. QCanvas uses a standard window-based graphical user interface (GUI), providing multiple windows to comparatively visualize patterns of various combinations of similarity matrices and hierarchical clustering methods. This program provides quantitative trees for displaying clustering patterns and similarity measures together.
Heatmap optimization for pattern recognition
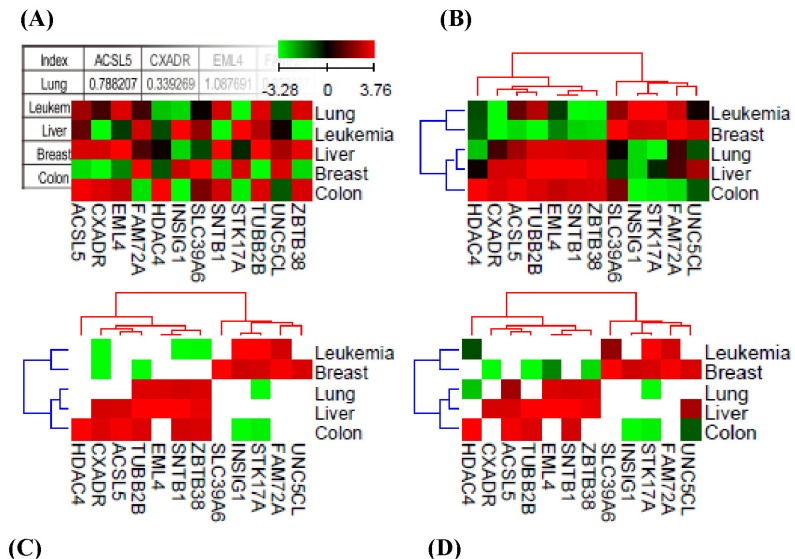
QCanvas software recognizes text-based data in a matrix format. For demonstration purposes, a small microarray gene expression dataset is included in the software package and can be downloaded from the website (http://compbio.sookmyung.ac.kr/~qcanvas). Once the input data are imported into the QCanvas window, a heatmap of the non-clustered data is displayed (Fig. 2A). The user can easily test various data-clustering and tree-building methods on the raw data and interactively select appropriate heatmaps with tree structures (Fig. 2B). The GUI provides various menu-based options to optimize the display of heatmaps, trees, and annotations. The colors, locations, and sizes of the trees and the annotations can be customized in a flexible manner. The scale and color scheme of the heatmaps can also be adjusted in an interactive window. The node colors can be customized for positive, negative, missing, or zero values. The color contrast between nodes can also be interactively adjusted. The overall vertical or horizontal size of a component of a figure can be customized and saved in postscript format for a high-image quality.
Data filtering for the selection of major markers
Heatmaps that are based on data clustering display the overall profiles of the experimental values for the given samples. QCanvas provides a data-filtering option to selectively display data nodes satisfying a given threshold. In the example shown in Fig. 2C, data points with a 2-fold change (increase or decrease) in gene expression are selectively displayed. In many cases, a dataset includes experimental values and statistical confidence levels together. The option for data filtering in QCanvas is useful for analyzing patterns in the experimental data that are statistically significant. One can filter the heatmap profiles using statistical confidence data that are included in a separate file. In the example shown in Fig. 2D, the gene expression data are filtered based on the p-values for the fold-change. QCanvas can import two separate files together for simultaneous data clustering and filtering. The GUI menu for data filtering enables the pattern analysis to be performed easily, without the need for manual data processing or the use of scripting languages.
Conclusion
This report introduces QCanvas, a program that provides a convenient and powerful interface for the pattern analysis of large-scale omics data. This program enables the user to conduct data clustering, data filtering, and graphics editing simultaneously on an integrated platform. These steps are typically performed on omics data, such as DNA (or protein) microarray data. All essential functionalities were integrated into the user-friendly interface of QCanvas. The simple and intuitive nature of this tool meets the practical needs of research scientists working on omics data who do not have expertise in bioinformatics approaches. The program is freely available with demo data and a step-by-step tutorial through the website (http://compbio.sookmyung.ac.kr/~qcanvas).