In silico detection and characterization of novel virulence proteins of the emerging poultry pathogen Gallibacterium anatis
Article information

Abstract
The pathogen Gallibacterium anatis has caused heavy economic losses for commercial poultry farms around the world. However, despite its importance, the functions of its hypothetical proteins (HPs) have been poorly characterized. The present study analyzed the functions and structures of HPs obtained from Gallibacterium anatis (NCTC11413) using various bioinformatics tools. Initially, all the functions of HPs were predicted using the VICMpred tool, and the physicochemical properties of the identified virulence proteins were then analyzed using Expasy's ProtParam server. A virulence protein (WP_013745346.1) that can act as a potential drug target was further analyzed for its secondary structure, followed by homology modeling and three-dimensional (3D) structure determination using the Swiss-Model and Phyre2 servers. The quality assessment and validation of the 3D model were conducted using ERRAT, Verify3D, and PROCHECK programs. The functional and phylogenetic analysis was conducted using ProFunc, STRING, KEGG servers, and MEGA software. The bioinformatics analysis revealed 201 HPs related to cellular processes (n = 119), metabolism (n = 61), virulence (n = 11), and information/storage molecules (n = 10). Among the virulence proteins, three were detected as drug targets and six as vaccine targets. The characterized virulence protein WP_013745346.1 is proven to be stable, a drug target, and an enzyme related to the citrate cycle in the present pathogen. This enzyme was also found to facilitate other metabolic pathways, the biosynthesis of secondary metabolites, and the biosynthesis of amino acids.
Introduction
Gallibacterium anatis (earlier known as Pasteurella anatis), which belongs to the family Pasteurellaceae, is an emerging disease-causing organism in poultry [1]. This bacterium has been isolated from various animals including chickens, turkeys, geese, ducks, pheasants, partridges, budgerigars, peacocks, cage birds, and wild birds [1-3]. There have been debates about whether this bacterium is pathogenic or nonpathogenic since it is found as a common part of the microbiota in upper respiratory tract and lower reproductive tract of healthy chickens [2,4,5]. However, increasing evidence indicates that G. anatis is associated with a wide range of pathological changes, leading to decreased egg production and lowered animal welfare in commercial poultry farms [6,7]. The disease is most likely to occur in intensively reared poultry farms (such as those raising broiler chickens) and incurs a high rate of mortality unless treated [1]. Therefore, determining the virulence factors of G. anatis would play an important role in proposing better control and prevention methods.
In recent years, numerous genomes have been sequenced with the help of next-generation sequencing technology and are available in public databases, such as that operated by the National Center for Biotechnology Information (NCBI) [8]. As a result, genomes with hypothetical proteins (HPs) have been deposited in sequence databases instead of experimentally confirmed facts due to their functional importance. Moreover, in some circumstances, because of limitations in illustrations (experimental validation techniques), expenditures, and the time required for the corresponding methodologies, whole-genome annotations have not been archived. Furthermore, the large amount of HPs in a genome naturally creates difficulties in data analysis [8]. This encourages in silico analysis, which utilizes and produces experimental information on HPs [9]. Establishing a structural and functional annotation for HPs may also play a significant role in elucidating protein pathways and cascades, helping to complete the currently approximate records on a variety of proteins [8,10]. Bioinformatics methods using discriminative algorithms and databases are the best approach to influence laboratory-based experimental procedures. Since these algorithms produce precise experimental results, they can be used to complete the functional and structural annotation of HPs [8].
The present study employed an in silico approach and predicted the functions of all HPs in the G. anatis reference genome (NCTC11413). Following identification, the physicochemical properties of the virulence-associated HPs were examined. Among them, a virulence protein within the pH range of 6–7 was identified as a drug target and analyzed for secondary structures, leading to the production of its first three-dimensional (3D) model by the ab initio method, and finally enabling the completion of its functional annotation. We believe that the present study provides a convenient methodology to analyze HPs and their functions in prokaryotic genomes.
Methods
Sequence retrieval and selection of HPs
The reference genome NCTC11413 was retrieved from the NCBI database. We observed that the genome consisted of 2,404 coding sequences. Upon analysis (https://www.ncbi.nlm.nih.gov/assembly/GCF_900450735.1/), 201 HPs were identified. All 201 HPs were separated from the genome using Geneious Prime version 2020.0.5.
In silico prediction of virulence factors, cellular processes, information/storage, and metabolism molecules
Identifying the functions of HPs plays a vital role in understanding a bacterium’s metabolic pathways and pathogenesis. The VICMpred tool (http://crdd.osdd.net/raghava/vicmpred/index.html) was employed for the identification of possible virulence factors, cellular processes, information/storage, and metabolism molecules among HPs from the reference strain NCTC11413. The VICMpred server is a support vector machine (SVM)–based method with the amino acid and dipeptide composition patterns of bacterial protein sequences [11]. The server provides an overall detection accuracy of 70.75%. At the end of the selection process, all virulence-associated HPs were selected for further characterization.
Physicochemical properties of virulence proteins
The physicochemical parameters of all virulence proteins were studied using Expasy's ProtParam server (https://web.expasy.org/protparam/), which was then used for computed theoretical measurements such as molecular weight, extinction coefficient, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). The extinction coefficient measures the amount of light that a protein can absorb at a certain wavelength. The instability index provides an estimation of the stability of a protein in a test tube, with a value of 40 indicating instability. The aliphatic index of a protein is described as the relative volume occupied by aliphatic side-chain amino acids. The GRAVY value for a peptide and/or protein is calculated as the sum of the hydropathy values of all of the amino acids, divided by the number of residues in the query sequence [12].
Subcellular localization and protein classification
It is a well-known fact that proteins present in the cytoplasm can serve as possible drug targets, while membrane proteins found on the surface are considered as vaccine targets. Thus, the subcellular localization of virulence proteins was predicted using the PSLpred online web server [it is a hybrid approach-based method that integrates PSI-BLAST and three SVM modules based on compositions of residues, dipeptides, and physicochemical properties] (http://crdd.osdd.net/raghava/pslpred/) and PSORT (a computer program for the prediction of protein localization sites in cells) (https://www.psort.org/). Moreover, the SignalP server (http://www.cbs.dtu.dk/services/SignalP/) was used to determine the presence of transmembrane helices and signal peptides [13-15]. This information is important for determining whether a protein is a membrane protein, secretory protein, or cytoplasmic protein. Following this, a virulence protein within the pH range of 6–7 was subjected to further characterization.
Functional domains, interaction network, and phylogenetic relationships of the protein
A virulence protein (WP_013745346.1) was examined to identify its precise functional domains using Pfam [16], HmmScan [17], Scanprosite [18], InterProScan [19], and SMART [20]. Additionally, the ProFunc server and STRING database were employed to understand possible functional interactions associated with the virulence protein [21,22]. Upon analysis, a phylogenetic tree was constructed using 14 other reference protein sequences in mega software version 7 [23] and the potential metabolic pathways were assessed using the Kyoto Encyclopedia of Genes and Genomes [24].
Secondary structure analysis
The SOPMA server (https://npsa-prabi.ibcp.fr/NPSA/npsa_sopma.html) was used to predict the secondary structure (helix, sheets, and coils) of the virulence protein WP_013745346.1 [25]. In addition, the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) was utilized to confirm the results achieved from SOPMA [26].
Homology modeling of the HP
The possible 3D structure of the virulence protein (WP_013745346.1) was created by an alignment approach on the SWISS-MODEL protein structure homology modeling server (https://swissmodel.expasy.org/) and the Phyre2 server [12,25].
Quality assessment of the 3D model and visualization
The early structural model of the achieved protein was checked for mistakes in the 3D structure using the ERRAT and Verify3D programs (https://servicesn.mbi.ucla.edu/) for structural examination and confirmation of protein modeling [26,27]. Finally, the PROCHECK structural evaluation server was used to assess the quality of the 3D structure [28]. The visualization of creating the model was accomplished by Geneious Prime version 2020.0.5.
Submission of the model to a protein model database
The protein model was created and successfully submitted to the Protein Model Database for the virulence protein (WP_013745346.1).
Results and Discussion
In silico prediction of virulence factors, cellular processes, information/storage, and metabolism molecules
Initially, by using the reference genome NCTC11413 we identified 201 uncharacterized HPs for G. anatis. These 201 HPs were analyzed using the VICMpred tool to understand their functional attributes. In return, the HPs were divided into four groups: 119 for cellular processes, 61 for metabolism molecules, 11 for virulence, and 10 for information/storage. The 11 virulence proteins identified in the present study were subjected to further characterization (Supplementary Tables 1 and 2).
Physicochemical properties, subcellular localization, and protein classification of the virulence proteins
The novel virulence proteins identified had 333–3,033 nucleotides and 111–1,011 amino acids (Table 1). Among the virulence proteins, the highest extinction coefficient (1.4 × 105) was observed in the HP WP_043885272.1. The instability index indicated that WP_013745190.1, WP_013746187.1, and WP_013746977.1 were unstable proteins, whereas the others were stable proteins (Table 2). Following this, the prediction of subcellular localization using PSORT indicated that 10 virulence proteins were cytoplasmic membrane proteins, whereas PSLpred showed three cytoplasmic proteins, two extracellular proteins, and six outer or inner membrane proteins. It is well known that proteins in the cytoplasm are possible drug targets, while membrane proteins are considered to be vaccine targets [29]. Cytoplasmic proteins play a crucial role in metabolic pathways that are critical to the survival of the pathogen inside the host organism [30]. Therefore, the three proteins detected in the cytoplasm (from both PSLpred and PSORT servers) can be used as drug targets, whereas the six membrane proteins could be used as vaccine targets against the present pathogen (Table 3). Moreover, the virulence-associated HP named WP_013746977.1 was identified as a signal peptide, whereas WP_013745329.1 was identified as a lipoprotein signal peptide. The protein structure of WP_013745346.1 (protein in the pH range of 6–7 and a possible drug target).
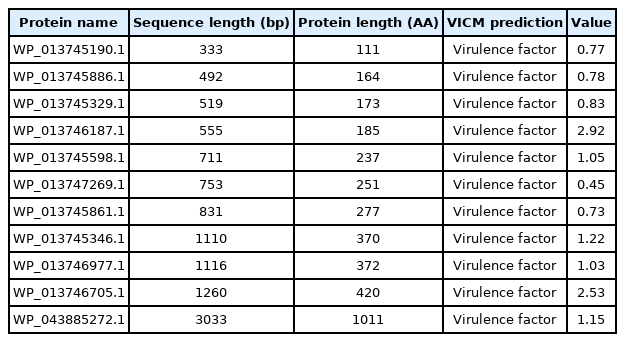
Novel virulence proteins identified in the Gallibacterium anatis NCTC11413 genome by the VCMPred online server
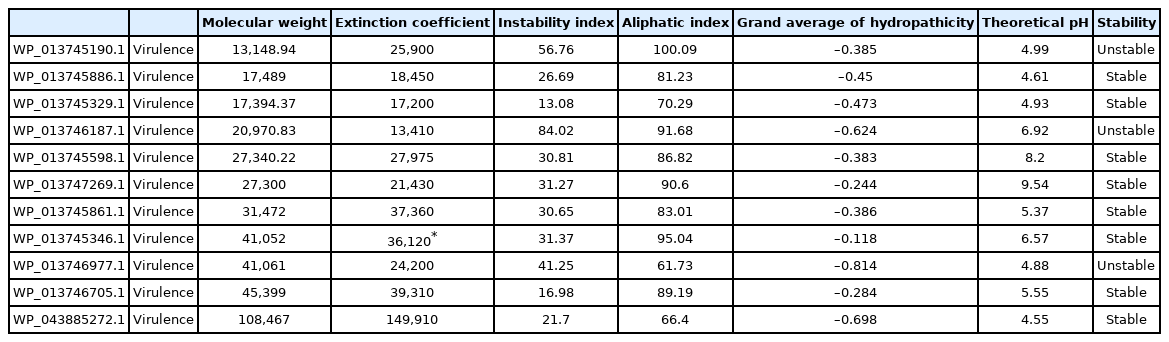
Physicochemical properties of 11 virulence proteins of Gallibacterium anatis with the ProtParam online server
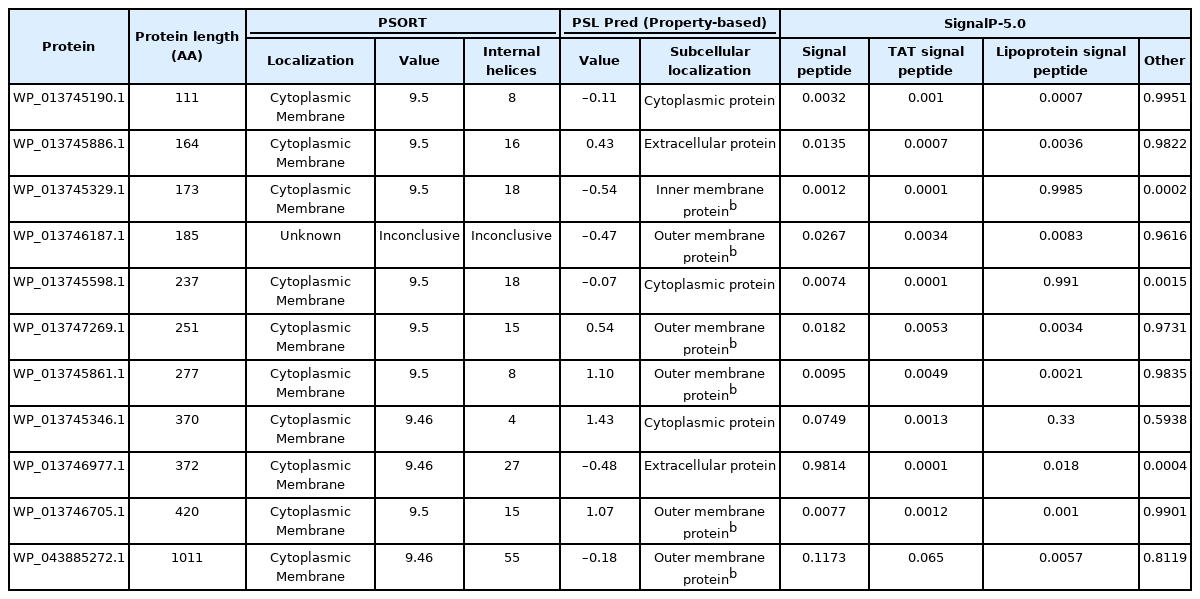
Subcellular localization and classification of 11 virulence proteins of Gallibacterium anatis with PSORT, PSLpred, and SignalP-5.0 online servers. (unknown: the PSORT server predicted the same values for all the functional categories)
Functional annotation of the virulence protein WP_013745346.1
All five employed tools indicated that the virulence protein WP_013745346.1 is an enzyme (known as methylcitrate synthase) primarily associated with the citrate cycle (Supplementary Table 3). ProFunc analysis indicated it is related to both biological processes (cellular process [74.47], cellular metabolic process [74.47], metabolic process [74.47], cofactor metabolic process [56.61]) and biochemical processes (catalytic activity [74.47], transferase activity [73.74], transferase activity/transferring acyl groups [47.82], transferase activity/ transferring acyl groups/acyl groups converted into alkyl on transfer [41.91]). The STRING protein network analysis suggested that the virulence protein WP_013745346.1 is associated with other functional proteins such as acsA and prpB (Fig. 1). The phylogenetic analysis with other reference sequences revealed that WP_013745346.1 is similar to other bacterial methylcitrate synthases (Supplementary Fig. 1). Moreover, this virulence protein was related to the biosynthesis of secondary metabolites, microbial metabolism in diverse environments, carbon metabolism, the biosynthesis of amino acids, and glyoxylate and dicarboxylate metabolism (Supplementary Fig. 2). This enzyme is a key determinant of propanoate degradation in micro-organisms [31]. Previous studies also suggested that the presence of methylcitrate synthase, malate synthase, and methylcitrate dehydrate is essential to the intercellular growth, metabolism, and virulence of bacteria, such as Mycobacterium tuberculosis [32]. Moreover, methylcitrate synthase in fungi, such as Aspergillus fumigatus, has the potential to detoxify propionyl-CoA, which is a byproduct of protein utilization; therefore, it can be used as a novel drug target [33].
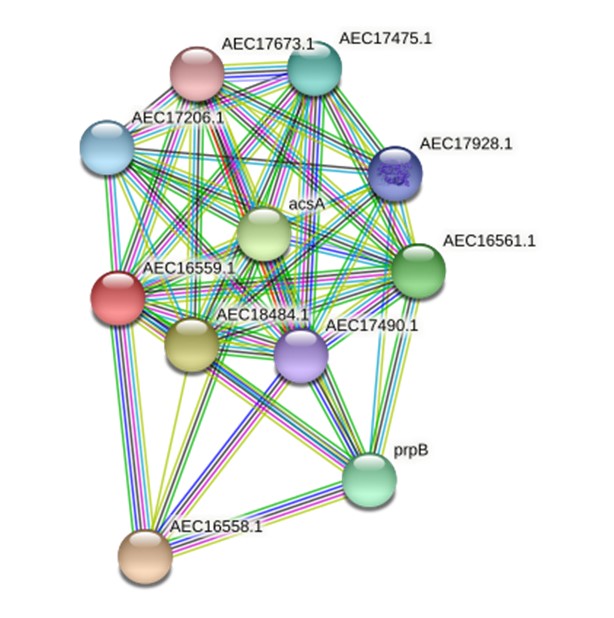
A STRING database search was carried out to identify a possible functional interaction network of the virulence protein WP_013745346.1 (AEC16559.1). The identified functional protein partners with the corresponding scores were as follows: AEC16558.1 (0.997), AEC18484.1 (0.966), acsA (0.962), AEC16561.1 (0.952), prpB (0.948), AEC17475.1 (0.920), AEC17206.1 (0.908), AEC17928.1 (0.907), AEC17490.1(0.877), and AEC17673.1 (0.841).
Secondary structure of the virulence protein WP_013745346.1
The secondary structure of the protein WP_013745346.1 was predicted using the SOPMA server. Alpha helices were found to be the most predominant (53.93%), followed by random coils (32.25%) and extended strands (8.40%). Beta-turns accounted for 5.42% of the structure of this protein. The predicted secondary structure for the virulence protein WP_013745346.1 from the PSIPRED server was also similar (Fig. 2).

The secondary structure of the virulence protein WP_013745346.1 was predicted using the PSIPRED server.
Homology modeling of the virulence protein WP_013745346.1
G. anatis infections have been reported in recent years at intensively reared poultry farms. However, its virulence-related factors have not been fully elucidated so far. Previous studies have demonstrated that the identification of virulence proteins from HPs plays a significant role in understanding its pathogenesis [28,29]. In silico analysis can help determine the biological functions of virulence proteins [8]. These predictions can be further strengthened by determining the 3D structures of virulence proteins using homology modeling. Homology modeling identifies the 3D structure of a selected protein sequence through alignment to one or more proteins of other known structures [34]. To perform homology modeling, the query sequence of virulence protein WP_013745346.1 was submitted to the SWISS-MODEL server. The server performed a BLASTP search for the respective protein sequence to identify templates associated with homology modeling. The highest identity of 36% observed for this virulence protein indicated that WP_013745346.1 is novel and no similar template structure is currently present in other databases. Following this, we determined the 3D structure of the virulence protein WP_013745346.1 by the ab initio method through the Phyre2 server, which gave 99.8% confidence in the model (Fig. 3). A comparative analysis of C. burnetii and M. tuberculosis methylcitrate synthases to WP_013745346.1, showed a common structural domain (citrate synthase, C-terminal domain), cellular location (cytoplasm), and molecular functions (Supplementary Fig. 3).
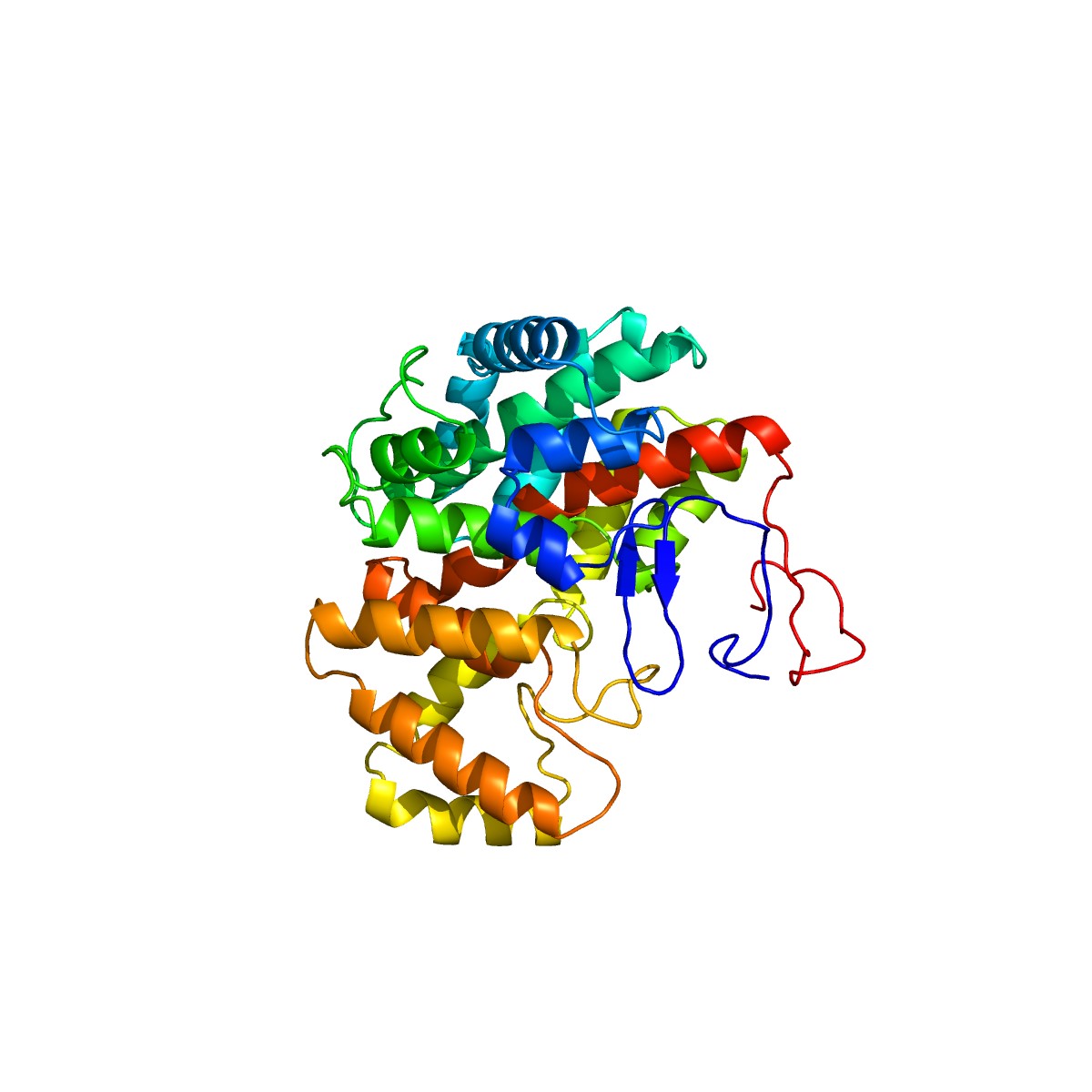
Three-dimensional structural analysis and visualization of the virulence protein WP_013745346.1 with Geneious Prime version 2020.0.5.
Quality assessment and visualization
The reliability of the created protein model was assessed using the ERRAT server, which analyzes the statistics of non-bonding interactions between diverse atom types, based on characteristic atomic interactions. The overall quality factor was found as 96.096%, which was sufficient to use this model. As shown by the Verify3D program, the results indicated 86.60% of residues had an average 3D (atomic model) – 1D (amino acid) score ≥0.2, meaning that this structure was compatible and genuinely good. Next, validation through a Ramachandran plot analysis showed that the distribution of φ and ψ angles in the model were within the limits (Table 4), 90.2% of the residues are in the most favored region of the plot, indicating that the model was valid (Fig. 4).

Ramachandran plot statistics of the virulence protein WP_013745346.1 predicted by the PROCHECK server
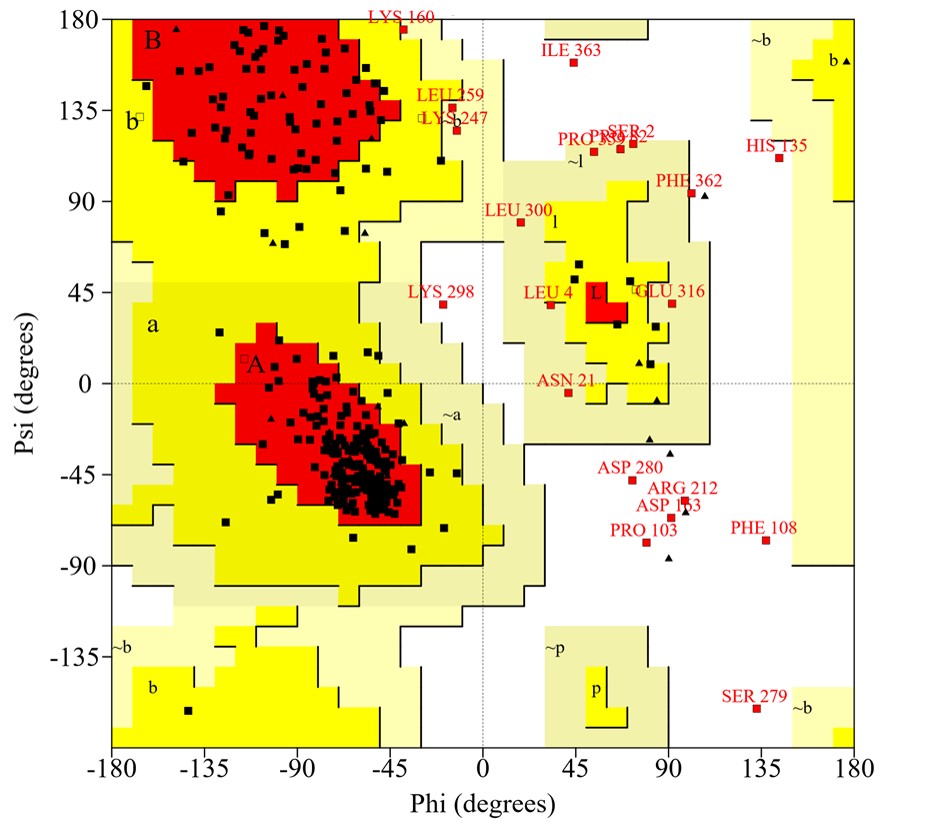
Ramachandran plot of the modeled structure for the virulence protein WP_013745346.1 validated by the PROCHECK program.
Data availability
The model created for the virulence protein WP_013745346.1 is currently available in the protein model database under reference number-PM0083267.
Summary
The present study aimed to characterize the HP functions of the emerging poultry pathogen G. anatis, as well as to create the first 3D structure and propose possible functions of the virulence protein WP_013745346.1. We observed that this novel protein is a stable cytoplasmic protein and functions as an enzyme in the citrate cycle. This protein was observed to be central to several other metabolic pathways. Therefore, the novel virulence protein studied here may have a significant impact on the pathogenesis of G. anatis.
Notes
Authors’ Contribution
Conceptualization: LGTGR, CWRG. Data curation: LGTGR, CWRG. Formal analysis: LGTGR, CWRG. Methodology: LGTGR, PSDA. Writing - original draft: LGTGR. Writing - review & editing: LGTGR, PSDA.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Supplementary Materials
Supplementary data can be found with this article online at http://www.genominfo.org.
Functional characterization of 201 hypothetical proteins of the poultry pathogen Gallibacterium anatis
Novel proteins identified from poultry pathogen Gallibacterium anatis associated with virulence, metabolism, Information and cellurer processes.
Functional domain prediction by five web servers for the virulence protein WP_013745346.1
Phylogenetic relationships of the virulence protein WP_013745346.1 with other reference protein sequences. (The evolutionary history was inferred using the neighbor-joining method. The bootstrap tree was inferred from 1,000 replicates and taken to represent the evolutionary history. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. The analysis involved 15 amino acid sequences. There was a total of 262 positions in the final dataset. The evolutionary analysis was conducted in MEGA7).
Potential metabolic pathways associated with the virulence protein WP_013745346.1 by KAAS (KEGG automatic annotation server analysis).
Comparative analysis of the 3D structure, structural domains, cellular location, and molecular functions of reference methylcitrate synthases to WP_013745346.1