Introduction
Lactobacillus are a group of Gram-positive, rod-shaped, microaerophilic, non-spore-forming, lactic acid–producing bacteria [1], they are the natural and significant inhabitants of gastrointestinal tract of humans, as well as they are known to constitute a major part of the oral and vaginal microbiome [2-5]. Lactobacillus are the most common probiotics found in fermented food products, and the awareness of probiotic benefits is evolving more quickly. Commercially available Lactobacillus probiotic strains help to restore the microbiota of imbalanced gut caused due to antibiotic treatments; however, the pathogenicity and efficacy of potential probiotics have to be assessed for safety. Here, we report the whole genome sequence of commercially potent probiotic Lactobacillus strains: Lactobacillus acidophilus UBLA-34, Lactobacillus paracasei UBLPC-35, Lactobacillus plantarum UBLP-40, and Lactobacillus reuteri UBLRU-87.
Lactobacillus strains were isolated from serially diluted fermented foods under anaerobic conditions at 37℃ using MRS (deMan, Rogosa, and Sharpe) agar, the pure isolated colonies were cultured using MRS broth, the cells were harvested for DNA isolation with the phenol-chloroform extraction method, followed by 16S rRNA gene amplification (using the primers 27F and 1429R) [6], the strains were confirmed by PCR amplicons sequencing and phylogenetic analysis. High molecular weight genomic DNA of the identified strains was isolated by the above-described method, DNA fragments of 300- to 400-bp size were generated by ultrasonication, fragmented DNA was used to prepare a paired-end sequencing library with a Nextera DNA Flex Library preparation kit (Illumina, San Diego, CA, USA) and sequencing was performed on an Illumina NextSeq 500 System (Illumina).
A total of 2,735,462 (420× genome coverage), 2,213,461 (218× genome coverage), 2,337,040 (214× genome coverage), and 1,641,982 (270× genome coverage) paired-end reads were generated for L. acidophilus UBLA-34, L. paracasei UBLPC-35, L. plantarum UBLP-40, and L. reuteri UBLRU-87, respectively. The reads were quality filtered based on the Phred score using NGS QC Toolkit to remove low-quality sequences [7]. The quality-filtered paired-end reads were assembled to high-quality draft genomes (Table 1) by employing de novo genome assembler SPAdes version 3.11.1 [8] and the scaffolder SSPACE-standard version 3.0 [9].
The genomes were annotated using RAST [10] and the NCBI’s Prokaryotic Genomes Annotation Pipeline (PGAP) [11]. The genes were predicted and translated through the Prokaryotic Dynamic Programming Gene-finding Algorithm (Prodigal) program [12], following pathway identification with the Kyoto Encyclopedia of Genes and Genomes Automatic Annotation Server (KAAS) [13] (Table 2).
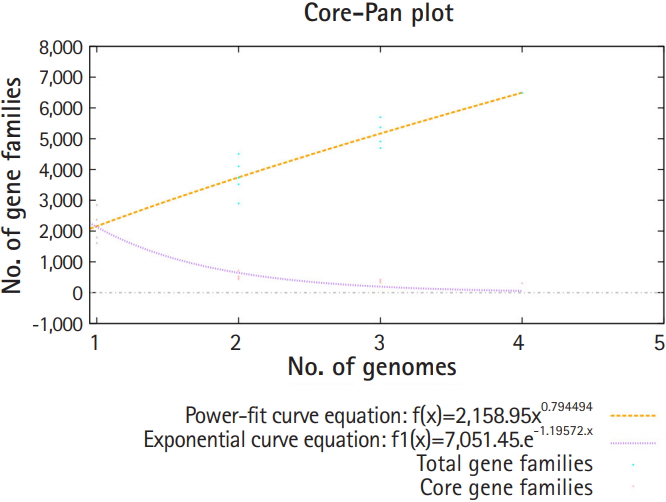
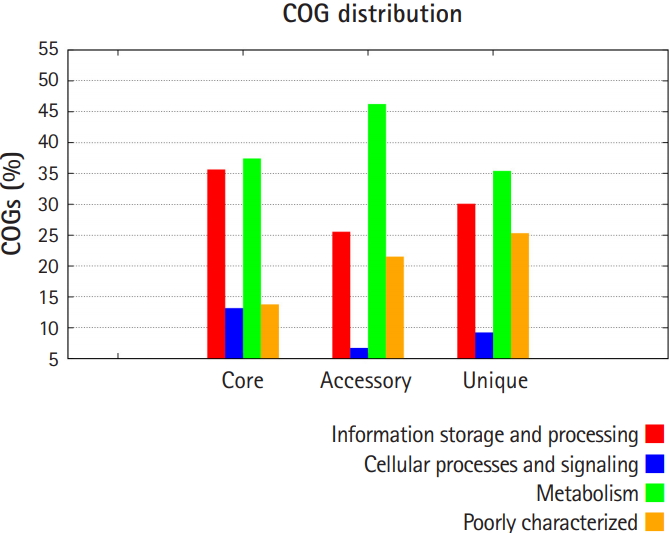
Pan-genomic analysis of Lactobacillus strains was performed to determine the conserved core and variable genes (Table 3) [14], the estimated pan-genome size was 6,487, and the parameter ‘b’ was calculated to be 0.794494 (Fig. 1), which confirms that the pan-genome is open. The highest number of new genes which contributed to the pan-genome was observed for L. plantarum UBLP-40 (Table 3). The highest part of the core genome of Lactobacillus genus was composed of genes related to metabolism, the second-highest contributing genes were related to information storage and processing, whereas the unique and accessory genes contained more amount of poorly characterized genes in comparison to core genome (Fig. 2). The phylogeny of core and pan-genome showed that L. reuteri shares the relatedness with L. plantarum, whereas L. paracasei is closest to L. acidophilus (Fig. 3).
All the four genomes of Lactobacillus strains were screened to determine the presence of genes encoding for putative virulence factors such as hemolysin BL, non-hemolytic enterotoxin NHE, enterotoxin T, cytotoxin T, and cereulide [15], antibiotic resistance [16], and plasmids [17]. None of the genomes (UBLA-34, UBLPC-35, UBLP-40, and UBLRU-87) showed the presence of putative virulence factor or antibiotic resistance encoding genes or plasmids or any antibiotic-resistant genes containing plasmids. Secondary metabolite producing gene cluster detection was performed for all the Lactobacillus strains, based on the hidden Markov model profiling of metabolite producing genes [18].
Lactobacillus acidophilus UBLA-34
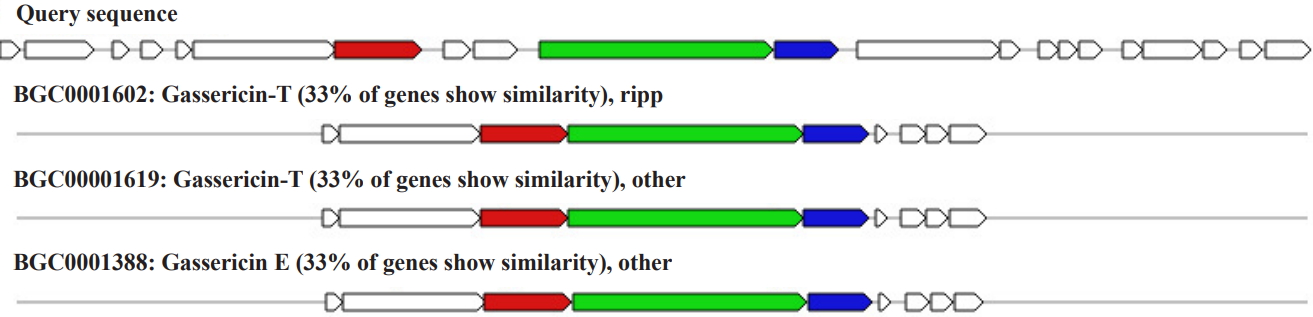
RiPP biosynthetic gene cluster was found in scaffold number 6 (location: 53,280–66,324 nt) consisting of seven genes encoding gassericin. The homologous gene cluster was mined from Lactobacillus gasseri LA327, gassericin T gene cluster Lactobacillus gasseri LA158 gassericin T gene cluster, Lactobacillus gasseri EV1461 gassericin E gene cluster with a 33% similarity (Fig. 4).
Lactobacillus paracasei UBLPC-35
Two bacteriocin biosynthetic gene clusters were found in scaffold number 1 (location: 21,360–44,300 nt and 85,659–97,824 nt), there was no significant similarity found with the known gene clusters.
Lactobacillus plantarum UBLP-40
First bacteriocin biosynthetic gene cluster was found in scaffold number 7 (location: 101,210–113,360 nt), whereas terpene biosynthetic gene cluster was found in scaffold number 12 (location: 77,136–92,747 nt), there was no significant similarity found with the known gene clusters.
Data Availability
The raw sequence reads have been submitted to the NCBI SRA and the whole-genome shotgun project has been deposited in DDBJ/EMBL/GenBank under the following accession numbers: Lactobacillus acidophilus UBLA-34: SRR7958229, RBHY00000000: the version described in this paper is version RBHY01000000, Lactobacillus paracasei UBLPC-35: SRR8382560, RCFI00000000: the version described in this paper is version RCFI01000000, Lactobacillus plantarum UBLP-40: SRR8382543, RDEY00000000, the version described in this paper is version RDEY01000000, Lactobacillus reuteri UBLRU-87: SRR8382542, RIAU00000000, the version described in this paper is version RIAU01000000.