Introduction
Viruses mutate faster than other microorganisms, and such mutations often lead to malignant infections in humans, animals, and plants. Therefore, it can be useful to develop methods to rapidly identify mutant viruses on the basis of International Committee on Taxonomy of Viruses (ICTV) taxonomy [1]. The function of a protein depends on its tertiary structure and alterations in protein tertiary structure leads to changes in protein function. Protein tertiary structure is determined by protein primary structure, which is comprised of the combination of amino acids. Therefore, from the genetic point of view, alterations in protein tertiary structure imply changes of the protein sequence in the coding region in the exons of the genome sequence. Thus, genetic mutations can alter the function and structure of protein and lead to disease. Therefore, in order to quickly detect the similarity of function at the emergence of new viruses, the final purpose is to analyze conserved domains, which can identify specific protein sequences for each virus. As the first application of this approach, we focused on norovirus, a positive single-strained RNA virus in Caliciviridae [2]. We extracted coding region (CDS) sequences in viral RefSeq GenBank and then apply the CDS protein sequences to the conserved domain database (CDD) (Table 1) [3]. Thereby, we assigned the meaningful annotation and selected specific protein sequences from domain tables generated by executing RPS-BLAST with the query of complete genome for each virus in Caliciviridae.
Methods
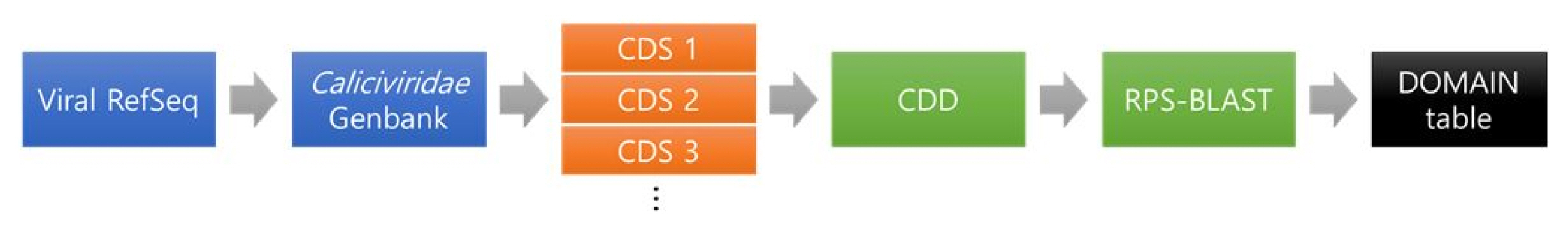
The method extracting specific protein sequence for each virus is shown in Fig. 1. The final goal is to choose conserved domains that can be utilized to identify genera in Caliciviridae.
ppub of RefSeq viral genomes
First, RefSeq raw data was collected from GenBank database in NCBI. As we know very well, NCBI provides GenBank files which include meta information and gene sequence information for each organism. However, the general GenBank files have problems that the information is duplicated because the same sequence information is submitted to NCBI by several institutes. This causes the increase of computation consumption. Thus, NCBI provides RefSeq GenBank files that minimized the sequence redundancy [4]. Thus, RefSeq data is appropriable to be used by reference sequences or standard sequences at executing a sequence alignment. In this study, the viruses data that are named by ICTV are only extracted from RefSeq genome sequences in NCBI database and then utilized in this study. The viral RefSeq sequences were parsed by the accession number starting with ŌĆ£NC_ŌĆØ notification implying the complete genome sequence. The information of viral RefSeq sequences can obtain from ŌĆ£viral 1.1ŌĆØ and ŌĆ£viral 2.1ŌĆØ (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/). There are norovirus species of 6 types: GI (NC_001959.2), GII (NC_029646.1), GIII (NC_029645.1), GIV (NC_0296471), GV (NC_008311.1), and primate norovirus (NC_031324.1).
Consistency verification for Caliciviridae
First of all, in order to verify consistency for whether the virus taxonomy named by ICTV was well classified or not, we compared all RefSeq sequences belonging to Caliciviridae by pairwise alignment sequence comparison (PASC) analysis [5], which provides assistance to search the sequences of the high similarity after pairwise comparing each complete genome in the genus level of Caliciviridae based on ICTV taxonomy, and by BLAST-based alignment method (blastn: nucleotide comparison). The NCBI RefSeq genomes of Caliciviridae exhibited a high homology by the PASC analysis were adequately matched to the classification defined by ICTV taxonomy. However, ŌĆ£NC_006875.1ŌĆØ showed the high similarity for Nebovirus (Table 2). It is consistent with the contention that ŌĆ£NC_006875.1ŌĆØ should be classified to Nebovirus [5].
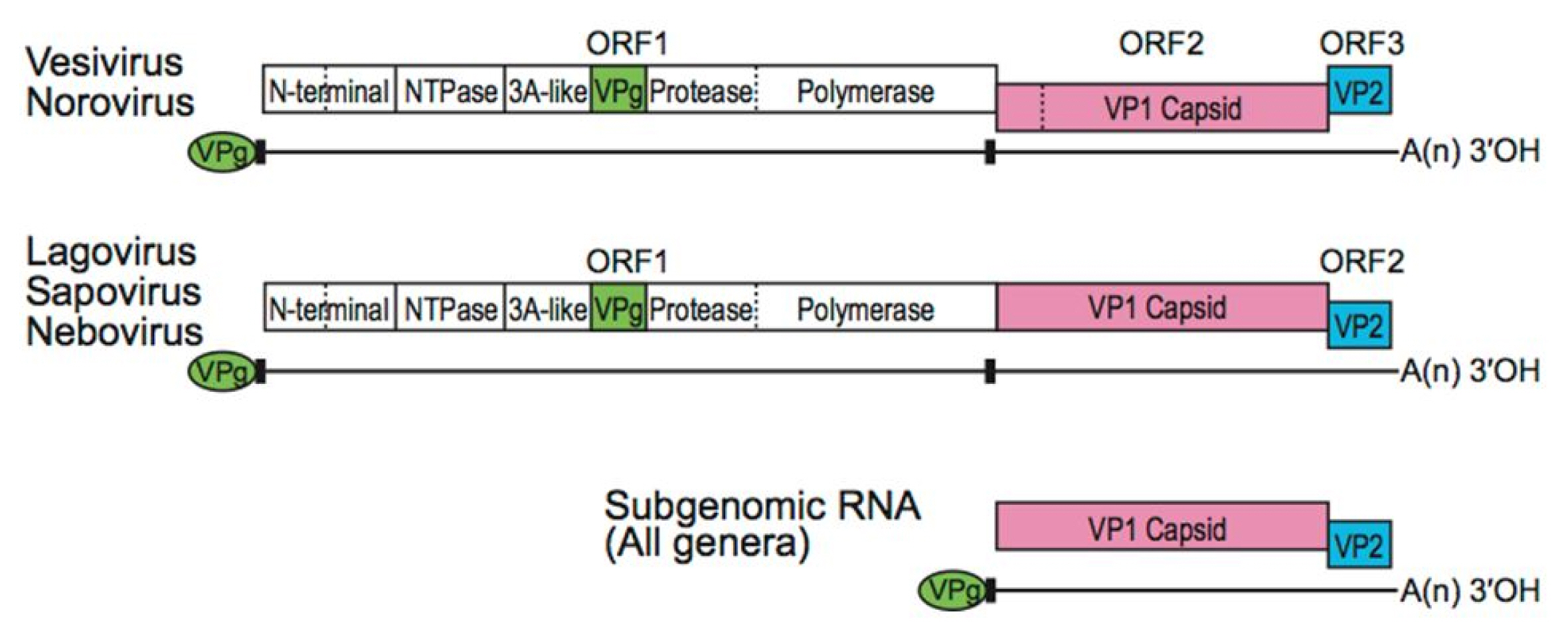
It is necessary to extract protein sequences from the region where gene expression appears. The extracted protein sequence can be split to open reading frames (ORFs). In viral GenBank, CDS sequences are specified with actual ORF sequences among such ORF fragments. An ORF is a continuous stretch of codons begun by a start codon (ATG) and terminated by a stop codon (usually UAA, UAG, or UGA). If transcription is terminated before the stop codon, an incorrect protein is produced. Caliciviridae have three ORFsŌĆöORF1, ORF2, and ORF3. ORF1 sequence is involved in the translation of non-structural polyproteins while ORF2 and ORF3 sequences are engaged in generating the major and minor capsid proteins, VP1 and VP2, respectively (Fig. 2) [6]. VP1 protein consists of two domains: P is split to P1, P2 and S. The P2 subdomain is considered with the region involved in cellular interactions and immune recognition [2].
Conserved domain search and annotation
The CDS sequences contain information about conserved domains. Domains are regarded as distinct functional and structural units, and the units can be repeated in similar protein structures [7]. The domains that are repeated in viruses in Caliciviridae can be utilized with a standard to identify the viruses in the genus level. By extracting the conserved domains, the final protein sequence that can be used to identify specific viruses can be selected. CDD was utilized to collect and assign annotations for the conserved domains [3].
By the technical method, we used reverse position-specific BLAST (RPS-BLAST). RPS-BLAST is the tool to search a protein sequence against a database of profiles, which are collected conserved domains. This tool is the opposite of PSI-BLAST searching a profile against a database of protein sequences. In this study, the database is composed as Table 1.
For the viruses in Calicivirdae, the conserved domains were almost defined by RPS-BLAST with Pfam database. Pfam database is generated by multiple sequence alignment and Hidden Markov models [8]. Generally, proteins structures are made by the combination of the domain units. To search common domain from each protein structure among the domains, there would provide some clues to identify the similar function and search the specific sequence for each protein. As we know, we cannot beg the question that the proteins have the similar functions even if protein sequences have the high similarity from the comparison between sequences. However, if we approach from the view of the conserved domain of protein structure as above the method (Fig. 1), we can find the specific domain for each protein and the domain can give help to define the specific sequences between the viruses in the same taxonomy.
Results and Discussion
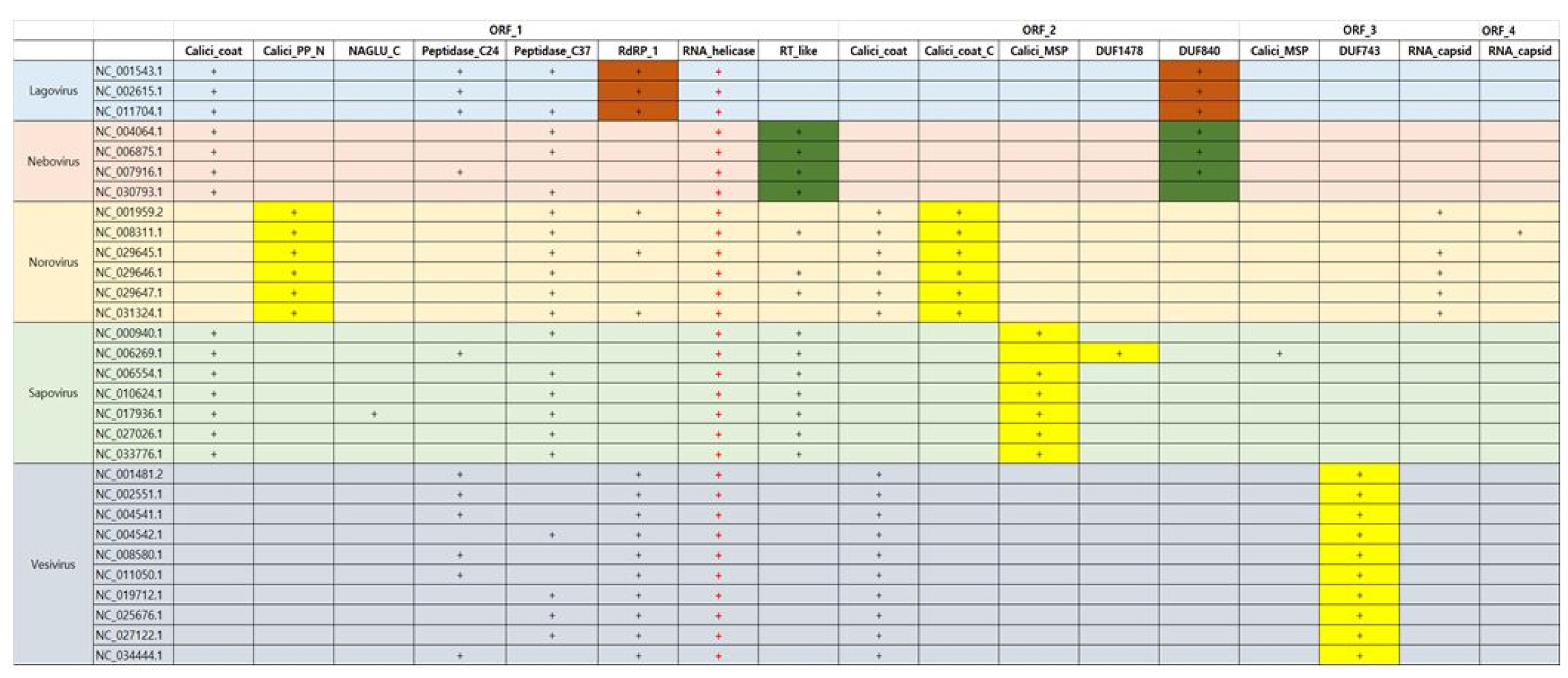
In Caliciviridae, all genera have RNA helicase in common as Fig. 3. In addition, RNA helicase exists in the type of single-strain RNA in common. As we know, RNA helicases have important roles in viral life cycles as well as in all cellular processes involving RNA. Vertebrate RNA helicases detects viral infections and causes the innate antiviral immune response. RNA helicases have been related with protozoic, bacterial and fungal infections such as neurological disorders, cancer, and aging processes. Thus, they can be utilized such as markers diagnosing symptoms and drug targets as well as antiviral and anti-cancer treatment [9]. In case of Norovirus, Calicivirus coat protein C terminal (Calici_coat_C) and viral polyprotein N-terminal (Calici_PP_N) appears unlikely as a specific domain in Caliciviridae, whereas the domain of Calicivirus minor structural protein (Calici_MSP) can be used as the specific proteins of Sapovirus. In here, Calici_PP_N is found at the N-terminus of non-structural viral polyproteins of the Caliciviridae subfamily. However, Vesivirus can be detected by protein of unknown function of DUF743 in CDD. In Caliciviridae, all viruses have Calicivirus coat protein (Calici_coat) domain in common. Lagovirus and Nebovirus contain Lagovirus protein of unknown function (DUF840), respectively. Lagovirus has only two species including rabbit hemorrhagic disease virus.
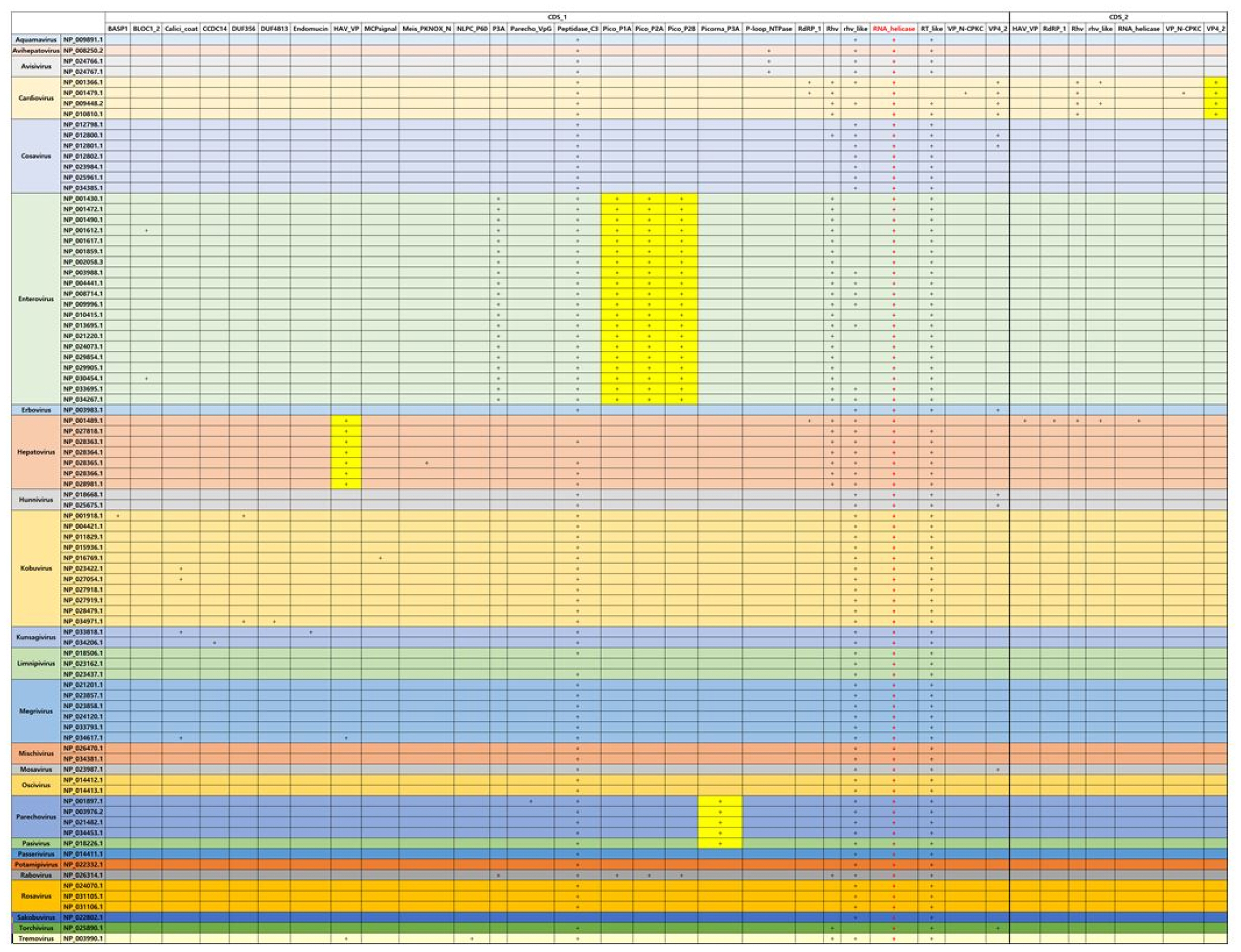
In case of Picornaviridae that is the same type with Caliciviridae, RNA helicase is also conserved in all genera in Picornaviridae. Specially, in case of Enterovirus, picornavirus coat protein (VP4) (Pico_P1A), picornavirus core protein 2A (Pico_P2A), which is a protease triggering polyprotein decomposition, and picornavirus 2B protein (Pico_P2B), which enhance membrane permeability, can be considered with the specific domains as compared with the other viruses in Picornaviridae. However, the E-value of Rabovirus is higher than that of Entertovirus even if the domains of Enterovirus seems to have a similar pattern with that of Rabovirus. Hepatovirus has Hepatitis A virus viral protein VP (HAV_VP) as the specific domain. This protein is found in hepatitis A viruses and targeted to the liver. Cardiovirus has unlikely the subunit of viral protein VP4 (VP4_2) (Fig. 4).
Overall, in terms of the methodology searching the specific protein sequences in order to identify the virus, the viruses could be classified by RPS-BLAST search with CDD and CDS sequence queries, which are parsed from viral RefSeq GenBank data. The viruses of Caliciviridae and Picornaviridae of single-strained RNA type are conserved to RNA helicase in common, which plays important role for viral infections detection and grasping the innate antiviral immune response. Calici_coat_C, Calici_MSP, and protein of unknown function (DUF743) could be considered with the specific protein sequence of Norvirus, Sapovirus, and Vesivirus in Caliciviridae, respectively. In addition, in the case of Picornaviridae, which is the same type with Caliciviridae, Pico_P1A, Pico_P2A, and Pico_P2B could regard as the specific protein sequence of Enterovirus. Therefore, suppose the method of Fig. 1 is applied to all viruses, the specific protein domain of each virus could be determined or compared by conserved domain searching with RPS-BLAST. It would provide useful clues for searching the specific protein sequences. If the specific protein sequences are defined, it could be converted to gene sequences. It would be utilized usefully to find viral bio-marks based on functional structure information of protein domain as well as used as classification keyword.