Introduction
Mutation rates include genetic changes such as single nucleotide polymorphisms (SNPs), copy number variations (CNVs), insertion/deletions (indels), microsatellites and minisatellites. Specifically, the subset of the total mutation rate caused by SNPs is called the substitution rate. Rate of point mutation can be determined indirectly by estimating neutral substitution rates [1]. In spite of the importance of substitution rates, there have been few investigations to date. We estimated the substitution rates of Hanwoo and Holstein cattle using the concept of the hidden substitution factor (HSF). The vast number of SNPs in next-generation sequencing (NGS) require the use of HSF to estimate the substitution rate. We defined the HSF as an independent unit for substitution rate in any lineage. For instance, the initial founder number is the HSF if the effective population size has been maintained as a constant. Otherwise, HSF is related to both founder number and effective population size. Using HSF, we estimated the genetic substitution rates of the genes of the Hanwoo and Holstein breeds in Korea.
Kumar and Subramanian [1] estimated the substitution rate in the mammalian genes using amino acid degenerate sites and evolutionary distance. Lee et al. [2] obtained the nonsynonymous SNPs, splice-site variants and coding indels in the Bovine genome including Hanwoo. They used whole-genome resequencing. Melka et al. [3] used Bovine SNP 50 beadchip to identify genomic differences between Hanwoo and Holstein. In our analysis, we used whole-genome sequencing to estimate substitution rates of Hanwoo and Holstein and identified genomic differences between Hanwoo and Holstein using gene ontology (GO) analysis. Our novel approach was to use intraspecies substitution rate estimation by inserting HSF into the evolutionary distance. HSF of Hanwoo is closely related to the initial found number that was caused by migration into Korean peninsula. We hypothesized that initial founder number was more than 1 [1-3].
Hanwoo designates the native, taurine type of Korean cattle. The breed originated approximately 4,000 years ago. The production of Hanwoo as the main beef cattle has occurred since the 1960s with the rapid growth of the Korean economy. The tasty beef from Hanwoo cattle is popular among Koreans and foreigners. Holstein-Friesian cattle represents the famous milk producing breeds. The holstein was introduced to Korea in 1902, and the Korean cattle industry has developed rapidly since 1960 [4-6]. Hanwoo and Holstein are very important in cattle industry in Korea.
We estimated the evolutionary distance of Holstein and Hanwoo using the number of SNPs, and HSF. Evolutionary distance refers to the cumulative change between DNA or protein sequences that were derived from a common ancestor. There were various methods to estimate it such as JC69 model, Kimura-2-parameter model (K2P), F81 model, HKY85 model. K2P used transition/transversion ratio (tr/tv ratio) to estimate the evolutionary distance. It is moderately complex model [7-10]. We assumed that the evolutionary distance had a linear relationship to HSF. The generation times of Hanwoo and Holstein were set to be 5 years. Then, the genetic substitution rates of the genes were calculated and those features were surveyed.
Methods
Data preparation
In this study, we used a whole-genome sequence data set consisting of 23 Hanwoo and 10 Holstein from NCBI Sequence Read Archive database (PRJNA210523, PRJNA210521, and PRJNA210519). We used fastQC software to perform a quality check on the raw sequence data [11]. Using Trimmomatic-0.32, we removed potential adapter sequences before sequence alignment [11]. Paired-end sequence reads were mapped to the reference genome (UMD 3.1.75) from the Ensemble database using Bowtie2 with default settings [12]. For downstream processing and variant-calling, we used open-source software packages: Picard tools (http://broadinstitute.github.io/picard/), SAMtools, and Genome Analysis Toolkit (GATK) [13, 14]. The “CreateSequenceDictionary” and “MarkDuplicates” Picard command-line tools were used to read the reference FASTA sequence to write a bam file containing a sequence dictionary, and to filter potential PCR duplicates, respectively. Using SAMtools, we created index files for the reference and bam files. We then performed local realignment of sequence reads to correct misalignments due to the presence of small insertions and deletions using the GATK “Realigner-TargetCreator” and “IndelRealigner” arguments. Additionally, base quality score recalibration was performed to get accurate quality scores and to correct the variations in quality associated with machine cycle and sequence context. For calling variants, GATK “Unified-Genotyper” and “SelectVariants” arguments were used with the following filtering criteria. All variants with (1) a Phred-scaled quality score of less than 30; (2) a read depth less than 5; (3) an MQ0 (total count across all samples of mapping quality zero reads) >4; or a (4) a Phred-scaled p-value using Fisher exact test of more than 200 were filtered out to reduce false-positive calls due to strand bias. We used the “vcf-merge” tools of VCFtools in order to merge all of the variant calling format files for the 33 samples [15].
The number of total SNPs was 37,484,886 and after using minor allele frequency; Holstein 0.1, Hanwoo 0.04) and Hardy-Weinberg equilibrium (p < 0.0001), the number of SNPs left in Hanwoo and Holstein were 12,626,097 and 8,636,673, respectively. The estimated transition/transversion ratio (tr/tv ratio) was calculated to be 2.24 using SnpEff [16].
The relationship between evolutionary distance and founder number
Evolutionary distance is linear to mutation rate and substitution rate [17, 18]. Evolutionary distance is proportional to the harmonic number of of sample size (a1) because the sample size can increase the number of SNPs. Theta represents the population mutation parameter (θ = 4Nμ). While Watterson’s theta estimator (θ ^ = # s e g r e g a t i n g s i t e s a 1
, where d is evolutionary distance, f is the HSF, a1 is the harmonic number of sample size, μ is the substitution rate, and t is the divergence time between the species and reference species.
HSF estimation via simulation
To estimate HSF, we simulated the average substitution rate of SNP chip data. We assumed the SNP number of general SNP chip data. The SNP chip data have 30,000‒50,000 SNPs in total, and the founder number can be assumed to be close to “1.” We simulated using the following parameters: the number of SNPs would be 30,000‒50,000 and the harmonic number of sample size would be 4‒6. Using Eq. (1), the expected value of the substitution rate was 3 × 10-10 to 5 × 10-10 (/bp/y). Using the result, we set the average substitution rate to be 4 × 10-10. This was a similar result to Roach et al. [22] who estimated the average mutation rate of human as 10-8 order per generation.
The HSF of Hanwoo and Holstein was estimated using Eq. (1). The evolutionary distances of the Hanwoo and Holstein population across chromosomes were calculated and then average HSF of Hanwoo (23 individuals) and Holstein (10 individuals) were estimated using the substitution rates of each chromosome.
Genetic substitution rate estimation of genes
After estimating HSF, we calculated each gene’s substitution rate in Hanwoo and Holstein populations. The bovine gene catalog was retrieved from the ensemble website (http://www.ensembl.org). The novel calculation was based on the number of SNPs inside each gene and used K2P model. We surveyed the genes that had the highest and lowest substitution rates. We compared the genetic substitution rates of Hanwoo with those of Holstein.
Results
HSF estimation
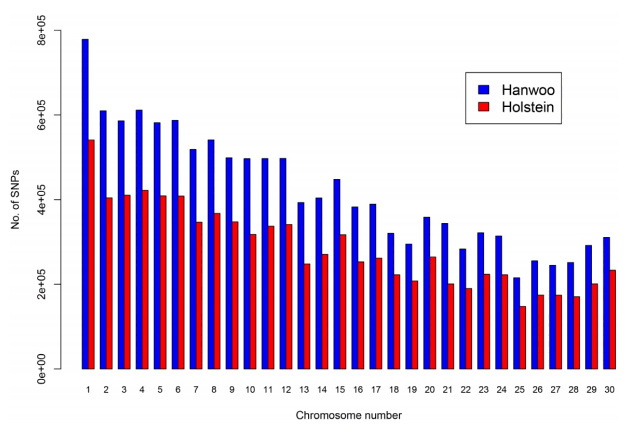
We estimated HSF of Hanwoo and Holstein. The HSF of Hanwoo and Holstein were 351 ± 46 (mean ± SD) and 279 ± 39 (mean ± SD), respectively. The Holstein HSF was not greater than that of Hanwoo because the Holstein HSF might pertain only to the history of Korean Holsteins. Fig. 1 shows the number of SNPs across chromosomes in Hanwoo and Holstein. The difference in the number of SNPs across chromosomes might be mainly due to the number of individuals i.e., 23 and 10 in Hanwoo and Holstein, respectively.
Substitution rate
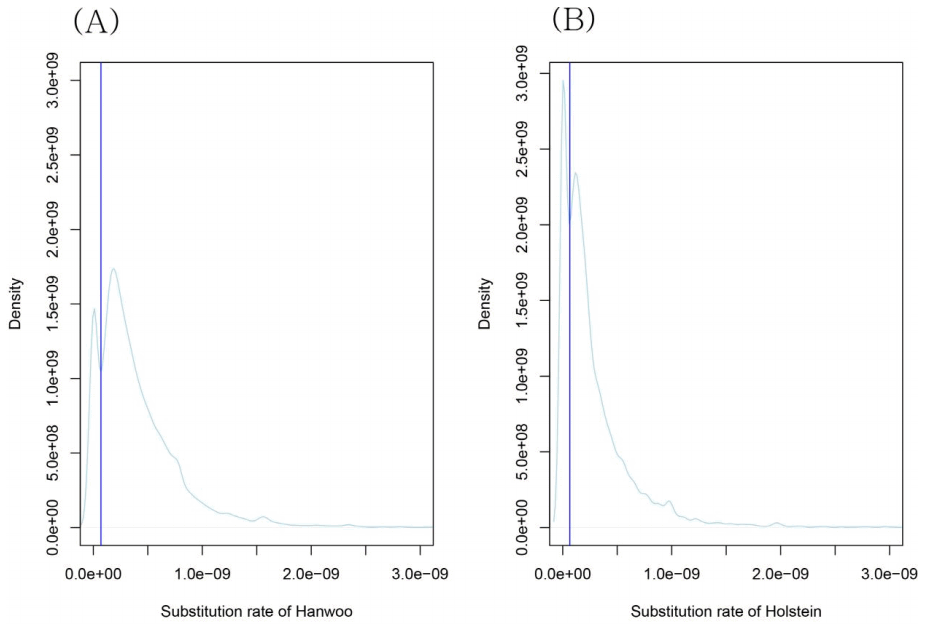
Fig. 2 shows the density of Hanwoo (Fig. 2A) and Holstein (Fig. 2B) substitution rates. The density of substitution rates showed the presence of a region in which the density showed significant decrease and increase. We might consider that this diving region differentiated the protected regions from unprotected regions. The substitution rates of the boundary regions in Hanwoo and Holstein were 7 × 10-11 and 6.5 × 10-11, respectively (/bp/y).
GO in Hanwoo and Holstein
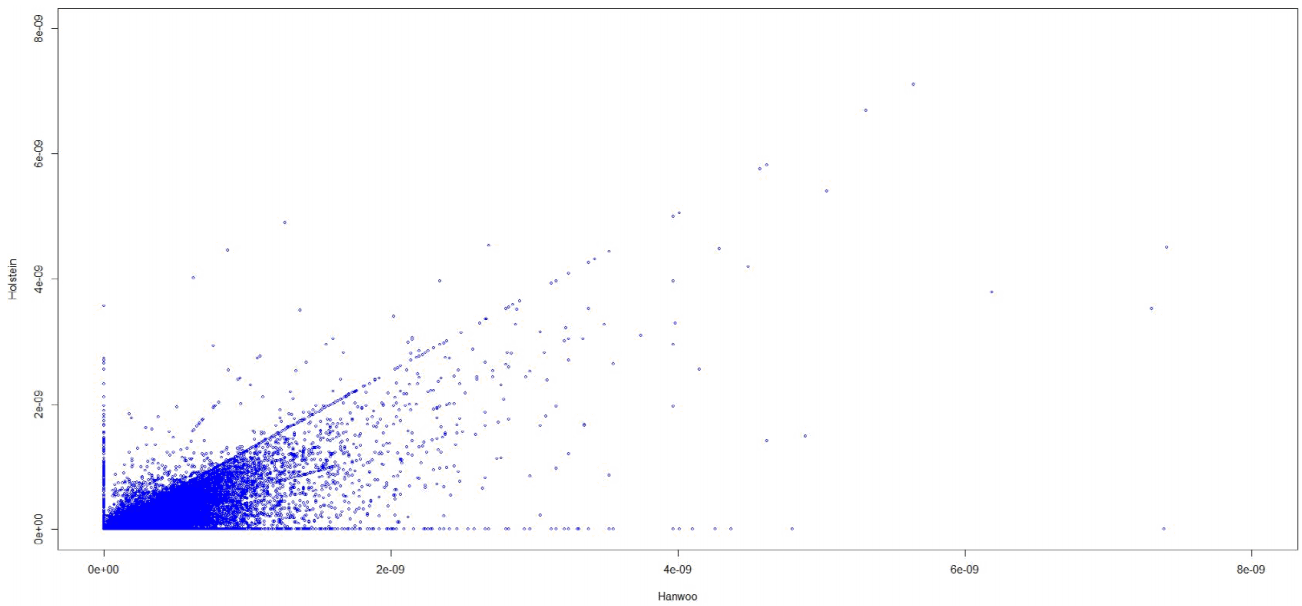
Table 1 shows the results of a DAVID analysis of GO terms for genes with low substitution rates (Hanwoo < 7 × 10-11; Holstein < 6.5 × 10-11). The criteria was substitution rates below diving regions. Most terms were regulation. It is obvious that regulatory genes would have low substitution rates. Table 2 shows GO terms of genes with different substitution rates between Hanwoo and Holstein. The major GO terms were related to blood and circulatory system. The criteria of genes was an over 20-fold difference in substitution rate between Hanwoo and Holstein (Fig. 4). Supplementary Table 1 shows the total genetic substitution rates and those ratios between Hanwoo and Holstein for protein-coding genes. The averages of gene substitution rates in Hanwoo and Holstein were 4.1 × 10-10 and 2.9 × 10-10. This was concordant with the simulation study of substitution rates (see Materials and Methods). We used the pseudocount 10-13 where the substitution rate in Holstein was estimated at zero to calculate the ratios.
Discussion
HSF estimation
The HSF can be viewed as the number of individuals requiring to explain the substitution rates present in a population. In SNP chip data, the HSF can be as close as 1. Founder effect can decrease nucleotide diversity and number of SNPs [23, 24]. Thus, smaller founder number can decrease the number of SNPs. Additionally, a population expansion after bottleneck events can increase the number of SNPs. Thus HSF should be estimated carefully, taking care of population history such as bottleneck event and population expansion.
The genes in Hanwoo and Holstein have average substitution rates on the order of 10-10. CNVs, microsatellites and minisatellites can also cause mutations. Therefore, it is very difficult to estimate the mutation rates with great accuracy. Instead, we focused on substitution rates which could be estimated using SNPs. The substitution rates on the order of 10-10 in Hanwoo and Holstein were similar to the human mutation rates [22].
The evolutionary distance can be assumed to be linear with HSF and the harmonic number of sample size (a1). Like Watterson’s theta estimator, the number of SNPs increases with sample size and thus we assumed that the evolutionary distance was linear with the harmonic mean of sample size (a1). Because HSF represents the independent substitution unit in the population, thus is linear with the number of SNPs in the population as Eq. (1).
Substitution diving region
The gene ontology (GO terms) analysis for genes with the lowest substitution rates in Hanwoo and Holstein showed that regulatory genes belong to the low substitution rate gene set. Regulatory genes might be protected genes. The existence of boundaries between the highest and lowest substitution rates showed that there were gaps in substitution rate between protected genes (lowest substitution rates) and unprotected genes (highest substitution rates). Such diving regions might not be the special case in Hanwoo or Holstein. This could provide evidence of differentiation between protected genes and unprotected genes.
GO results differentiating Hanwoo from Holstein
The differentiation of Hanwoo from Holstein was identified by comparing the genetic substitution rate at the gene level. In GO analysis of differentially substituted genes between Hanwoo and Holstein, the blood and circulatory system terms were most common. The disparate evolutionary patterns after domestication between Hanwoo and Holstein could be caused by these genes. Hanwoo, which mainly provides meat, and Holstein, which mainly provides milk, might evolve differently, and blood and circulatory system-related GO genes could play some role in the evolution of Hanwoo and Holstein.