Genomic Analysis of miR-21-3p and Expression Pattern with Target Gene in Olive Flounder
Article information
Abstract
MicroRNAs (miRNAs) act as regulators of gene expression by binding to the 3’ untranslated region (UTR) of target genes. They perform important biological functions in the various species. Among many miRNAs, miR-21-3p is known to serve vital functions in development and apoptosis in olive flounder. Using genomic and bioinformatic tools, evolutionary conservation of miR-21-3p was examined in various species, and expression pattern was analyzed in olive flounder. Conserved sequences (5’-CAGUCG-3’) in numerous species were detected through the stem-loop structure of miR-21-3p. Thus, we analyzed target genes of miR-21-3p. Among them, 3’ UTR region of PPIL2 gene indicated the highest binding affinity with miR-21-3p based on the minimum free energy value. The PPIL2 gene showed high expression levels in testis tissue of the olive flounder, whereas miR-21-3p showed rather ubiquitous expression patterns except in testis tissue, indicating that miR-21-3p seems to control the PPIL2 gene expression in a complementary repression manner in various tissues of olive flounder. Taken together, this current study contributes to infer the target gene candidates for the miR-21-3p using bioinformatics tools. Furthermore, our data offers important information on the relationship between miR-21-3p and target gene for further functional study.
Introduction
MicroRNAs (miRNAs) are groups of endogenous small, noncoding, single-stranded RNA molecules 20–24 nucleotides in length [1]. The miRNAs bind to the 3´ untranslated regions (UTRs) of its target mRNAs entirely or partly and they act as post-transcriptional inhibitors of gene expression [2, 3]. This event causes specific mRNA degradation or translational repression [4]. Each single miRNA could target numerous mRNAs, and a single mRNA can be targeted by several miRNAs, which allows to coordinate and control regulation of protein inhibition [5]. The miRNAs have played a role in a variety of biological processes such as cell proliferation, cell survival, DNA repair, and immune response [6]. Therefore, complex interactions involved in the regulation of gene expression have been well-established. At least up to 60% of protein-coding genes expressed in various species, and down-regulated more than 1,000 miRNAs [7].
The olive flounder Paralichthys olivaceus is one of the most important fish species that widely cultured in Korea, Japan, and China [8]. Recently, due to lots of consumption, its economic values have increased in the fish markets [9]. However, olive flounder is sensitive to infection of many bacterial pathogens [10] and they can trigger significant economic losses. Therefore, there has been an increase in studies that strive to understand the effects of bacterial infection in olive flounder via quantitative real-time polymerase chain reaction (qRT-PCR) [11]. Genetic variation and population structure among olive flounder populations are estimated using several molecular approaches. Those research results indicated proper yields and retaining genetic diversity [12]. Genetic variation is beneficial and important for the long-term survival of natural populations, as it ensures that a high level of fitness is maintained by giving populations the ability to adapt to changing environmental conditions [13]. Population genetic structure is influenced by the effects of gene flow, natural selection, genetic drift, and mutation [14].
According to the several functional studies, miRNA and their targets interaction in olive flounder indicated involvement in development, apoptosis, and metabolism [6, 15]. One of the representative studies is about the miR-17, and it is about metamorphic development of olive flounder that overexpressed miR-17 in FEC cells and suppressed Cdc42 expression due to miR-17 binds to 3´ UTR of Cdc42 gene. The declined metamorphic development means growth level of olive flounder also diminished [16]. Recently, studies have shown that 21 let-miR-7 precursors control metamorphosis stages from the larval to juvenile form in P. olivaceus. These let-7 miRNAs were shown in different positions of P. olivaceus genome and significantly expressed in adult tissues of olive flounder, but not expressed in embryonic development tissues, indicating that let-7 miRNAs could be associated with tissue development and metabolism process [17]. There is limited evidence between the miR-21-3p expression pattern and the quantitative expression levels of the target gene in various olive flounder tissues. Previous studies reported miR-21-3p function in different fields. For instance, some miRNAs have been found to function as oncogenic miRNAs, this can be observed in the study where miR-21-3p was found to be up-regulated in solid and hematological cancer tissues thus leading to the conclusion that miR-21-3p repress the expression of tumor suppressors like PTEN phosphatase and actin-binding protein tropomyosin I [18-20]. In addition, the set of miR-21 and its target mRNA, TGFBR2, were predicted as high binding affinity in seven types of solid tumor namely; lung, breast, colorectal, pancreatic, prostate, stomach, and bladder that indicated strong molecular interaction between miR-21 and TGFBR2 [21, 22]. The hsa-miR-21-3p was known as bipolar disorder (BP) dominant miRNA by verifying only increase of miR-21-3p in BP on fibroblast culture [23]. The miR-21-3p also upregulated expression in hearts of lipopolysaccharide-treated mice [24]. Due to down regulation of SH3 domain-containing protein 2 (SORBS2) is one of target genes of miR-21-3p and putative target of SORBS2 controls septic infection-associated cardiac dysfunction [24].
In the present study, the evolutionary conservation patterns of miR-21-3p in various species and miR-21-3p target genes were analyzed using bioinformatic tools. Also, expression patterns of miR-21-3p were analyzed in various tissues of olive flounder in relation to the target PPIL2 gene using qRT-PCR.
Methods
Total RNA isolation and cDNA syntheses
Total RNA was extracted from ten tissues of olive flounder (spleen, gill, muscle, kidney, intestine, stomach, liver, fin, brain, and testis) using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) depending on the manufacturer’s instructions. These RNAs were treated with RNase-Free DNase I (New England Biolabs, Beverly, MA, USA) to remove genomic DNA contamination. RNAs were separated by 1% agarose gel electrophoresis for analysis of the 28S and 18S RNA bands. The RNA concentration was quantified to 500 ng using a ND-1000 UV-Vis spectrophotometer (Nano Drop, Wilmington, DE, USA). ReverTra Ace qPCR RT Master Mix kit (TOYOBO, Osaka, Japan) was used for cDNA synthesis. Prior to use, a 1 in 50 volume of gDNA remover was added to 4× DN Master Mix and then each quantitated 500 ng RNA samples 6 μL denatured at 65℃ for 5 min. Then, 2 μL of 4× DN Master Mix was added to RNA in order to increase DNase I reaction and incubated at 37℃ for 5 min. After that, 2 μL of 5× RT Master Mix was added to 4× DN Master Mix. Reverse transcription response was performed by following steps; incubation at 37℃ for 15 min, 50℃ for 5 min, heating 98℃ for 5 min and stored the solutions at ‒20℃.
Quantitative real-time RT-PCR analyses
Quantified RNA samples were used for miR-21-3p expression and its target gene; HOXC9 3´ UTR expression analysis. HB miR Multi Assay Kit System I (Heim Biotek, Seongnam, Korea) was used to miRNA analysis that miR-21-3p reverse transcription (RT) and real-time PCR. First, 500 ng RNA reaction volume per each tissue need to 11 μL containing RNase free water. Then for synthesis of cDNA—HB_I RT Reaction Kit and its reagents was used in compliance with manufacturer’s instructions and reverse transcription polymerase chain reaction (RT-PCR) amplification was performed in a thermal cycler (Eppendorf, Hamburg, Germany) on terms of 37℃ for 60 min followed by incubation at 95℃ for 5 min for one cycle and then held at 4℃. Synthesized cDNA samples in various tissues olive flounder were stored at ‒20℃. Second, HB_I Real-time PCR Master Mix Kit from the HB miR Multi Assay Kit System I was followed by manufacturer’s protocols. All cDNA samples were amplified in triplicate to ensure reproducibility with a Rotor-Gene Q system (Qiagen, Hilden, Germany). Real-time PCR was initial denaturation for 15 min at 95℃, 45 thermal cycles of 95℃ for 10 s and 60℃ for 40 s and standard melting conditions of ramp ranging from 55℃ to 99℃ with 1℃ rise on each step. As a standard control, miRNA U6 was used as reference. Relative expression ratio of the target miR-21-3p (5’-CGACAACAGUCUGAAGGCUGUC-3’) to miRNA U6 were analyzed the comparative threshold method (2-ΔΔ Ct).
In case of target gene expression analysis, 7 μL of ddH2O, 10 μL of QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA), 1 μL of each forward (5´-TCGTGCA-TCTACCTCGACAG-3´) and reverse (5´-CCTCCTCCT-CCTTGGTTTTC-3´) primers and 1 μL of cDNA template. Real-time RT-PCR amplification for target gene and housekeeping gene were carried out 45 cycles of 94℃ for 10 s, 60℃ for 15 s, and 72℃ for 15 s. Melting curve was progressed for 30 s at 55‒99℃. All samples were amplified in triplicate and housekeeping gene was used universal glyceraldehyde 3-phosphate dehydrogenase (GAPDH) of sense primer (5´-GAAATCCCATCACCATCTTCCAGG-3´) and antisense primer (5´-GAGCCCCAGCCTTCTCCATG-3´). For statistical analysis, the expression data were measured as the mean ± standard deviation. Significant differences among various tissue samples were determined by the unpaired Student’s t test. The value of p < 0.05 was accepted as an indication of statistical significance. Differences less than p < 0.05 were considered.
Evolutionary conservation of miR-21-3p in various species
To identify miR-21-3p mature sequences in various vertebrate species, miRBase v19.0 (http://www.mirbase.org) was used to analyze. The miR-21-3p structure identification was performed by the mfold Web Server (http://mfold.albany.edu). The evolutionary conservation pattern of various species was examined by Evolutionary Conservation of Genomes (ECR browser; https://ecrbrowser.dcode.org/) in zebrafish, fugu, Tetraodon, chicken, opossum, rat, mouse, cow, dog, and chimpanzee.
The miR-21-3p target genes and binding site analyses
The search of target genes was performed by miRTarBase release ver. 6.0 (http://mirtarbase.mbc.nctu.edu.tw) and TargetScanHuman ver. 7.1 program (http://www.targetscan.org). Target genes were predicted and their interaction between human target genes and miR-21-3p was done by RNA hybrid ver. 2.1 program (http://bibiserv2.cebitec.uni-bielefeld.de/rnahybrid). Minimum free energy (MFE) score by binding affinity of miRNA and their target genes were also obtained in RNA hybrid program. TargetScan-Human program was used to verify binding regions of the miRNAs and their target gene 3´ UTR region. This program carries out whether target gene 3´ UTR sequences in several vertebrates. The mature miR-21-3p sequences were acquired from miRBase program matched with aligned 3´ UTR domain. Target genes of Gene Ontology (GO) for miR-21-3p was conducted through PANTHER (Protein Analysis Through Evolutionary Relationships) classification system (http://www.pantherdb.org/), which is related to the three functional categories: molecular function (MF), biological process (BP), and cellular component (CC).
Bioinformatic analyses of target genes
The interaction of target gene and its related genes was analyzed gene-gene interaction (GGI) by using Gene MANIA database (http://genemania.org/). In addition, to identify target protein, associated other proteins, STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) ver. 10.5 program for functional protein association network (https://string-db.org/) was used and commonly referred as protein-protein interaction (PPI). STRING database informed a degree of target and related protein similarity, as well as co-occurrence analysis among various species.
Results
Evolutionary conservation and genomic structure of miR-21-3p in various species
The conserved mature sequences of miR-21-3p in various species were analyzed by miRbase program. The 5´-CAGUCG-3´ sequences were highly conserved and indicated by shaded rectangles and marked with asterisk (Fig. 1). Using the ECR browser, evolutionary conservation of miR-21-3p was examined, indicating that miR-21-3p has been highly conserved from fishes to primates (Fig. 2). The structure of miR-21-3p was examined by mfold web server. The MFE value of miR-21-3p is ‒6.00 kcal/mol (Fig. 3).

Comparative analysis of miR- 21-3p in various species. The 5’-CAGUCG-3’ sequences were highly conserved. It was indicated by shaded rectangle and marked as asterisk.

Evolutionary conservation of miR-21-3p in various species by ECR browser.
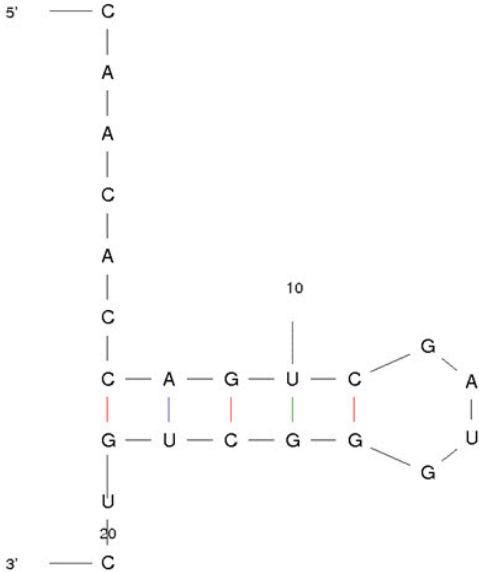
The stem-loop structure of miR-21-3p by mfold program.
Target genes analyses of miR-21-3p
All target genes were analyzed using GO by PANTHER Gene List Analysis system. One of the three categories BP associated to cellular process and metabolic process functions. MF and CC revealed that multiple genes were related to binding activity, catalytic activity, cell part, and organelle component, respectively (Fig. 4). Binding sites (5´-CGACUG-3´) of target genes for the miR-21-3p were analyzed by Targetscan program, which mainly appeared in species-specific genes (Table 1). MFE score from hybridization of seed region and binding site was almost less than ‒15 kcal/mol. The binding affinity between miRNA and 3’ UTR region of target genes seem to be strong in brown bat (KANK2), cat (SNAPC3), elephant (HN1), lizard (AIFM1), opossum (RELA), and rabbit (PPIL2) species as the average value of MFE was below ‒25 kcal/mol. Target genes generate stem-loop complexes with their respective miRNA at the seed region of miR-21-3p and the binding site of target gene (Fig. 5).
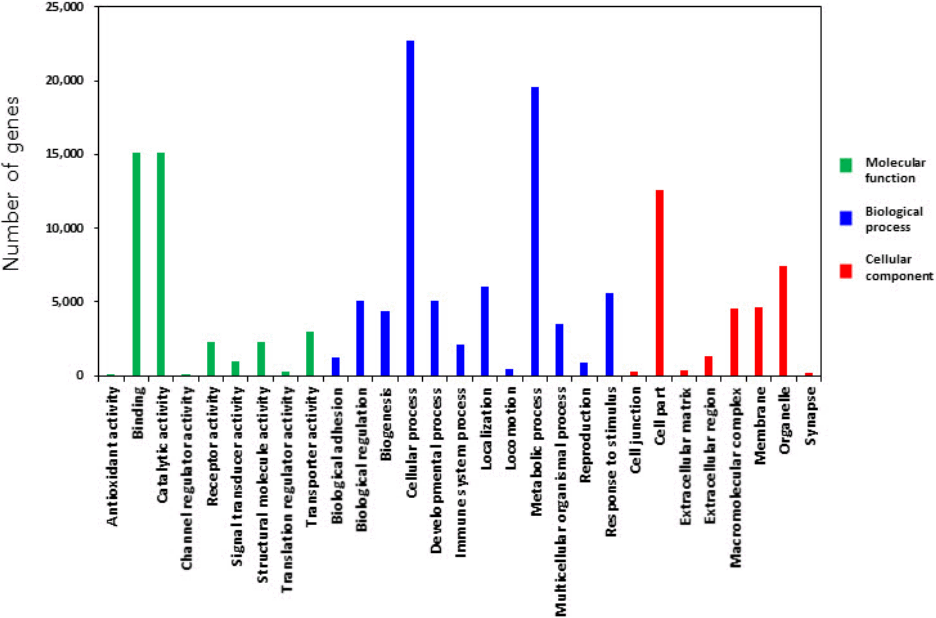
Gene Ontology analysis of target genes for the miR-21-3p by PANTHER program.

Target genes and their binding site for the miR-21-3p examined by Targetscan and minimum free energy (MFE) value
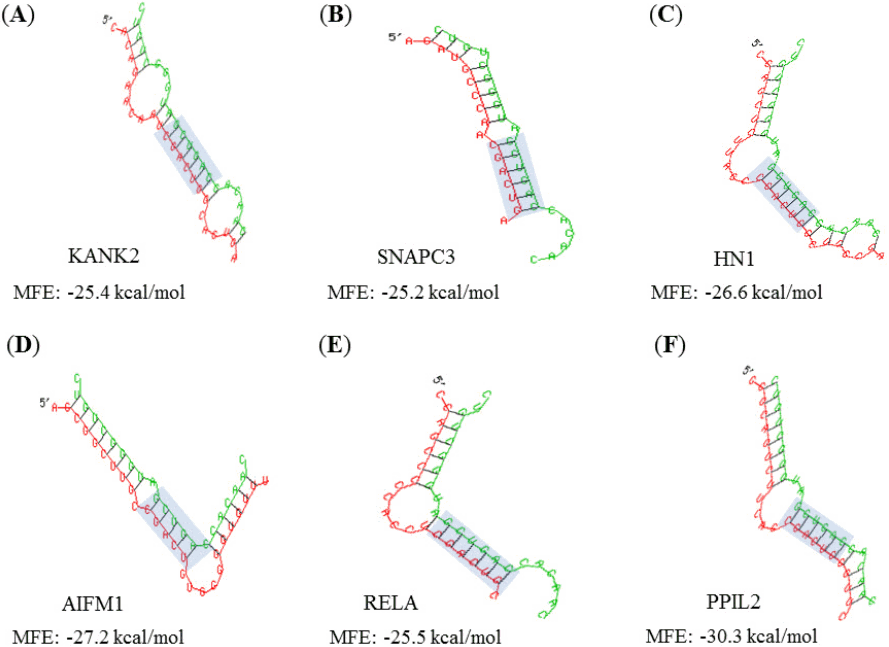
(A–F) The hybridization analyses between miR-21-3p and 3´ untranslated region (3´ UTR) of target genes. They show high level minimum free energy (MFE) score. Seed regions of miR-21-3p and binding sites of 3’ UTR region are indicated by shaded rectangles.
Bioinformatic analyses of target genes
PPIL2 is one of the target genes that have the highest MFE level and it was chosen to study the interaction with miR-21-3p due to its ‒30.3 kcal/mol for MFE score. GGI network was used to identify functional roles of target genes, where several genes such as PPM1F, DGCR2, ASCC2, LZTR1, and MVD were found to be co-expressed with PPIL2. The physical interaction function was associated with PRCC, PIM2, BSG, ZNF830, CRNKL1, and KANSL2. CWF19L1, a member of the CWF19 protein family, which controls the cell cycle was predicted to have a correlation with PPIL2. PPIL3, MED15, and DGCR14 showed co-localization ability each other (Fig. 6A). To confirm previous data, PPI network was examined by STRING database, and numerous proteins involved with PPIL2 (Fig. 6B). To understand in Fig. 6B, each circle of proteins means structure decision status, where large sized circles indicate prediction of protein structure. Most proteins that were expected to have a correlation with PPIL2 co-expression were experimentally determined. Also, PPIL2 showed gene neighborhood relationship except PRCC, PRPF19, SNW1, XAB2, and BVD31. Although most of proteins’ function has not been yet identified, TOP2A and TOP2B were known as DNA topoisomerase 2, controlling of topological states of DNA by transient breakage and subsequent rejoining of DNA strands. Proteins related to PPIL2 showed occurrence profile that have two features: similarity scale and clade coverage. The similarity scale in these presence/absence profile could predict protein interaction. Clade coverage means genome group were separated in the phylogenetic tree and two distinct colors indicate that the lowest and highest similarity observed within the clade. Most of proteins were conserved especially in primates compared with other populations (Fig. 7).

Gene-gene interaction by Gene MANIA (A) and protein-protein interaction by STRING program (B) for the target gene (PPIL2) of the miR-21-3p.
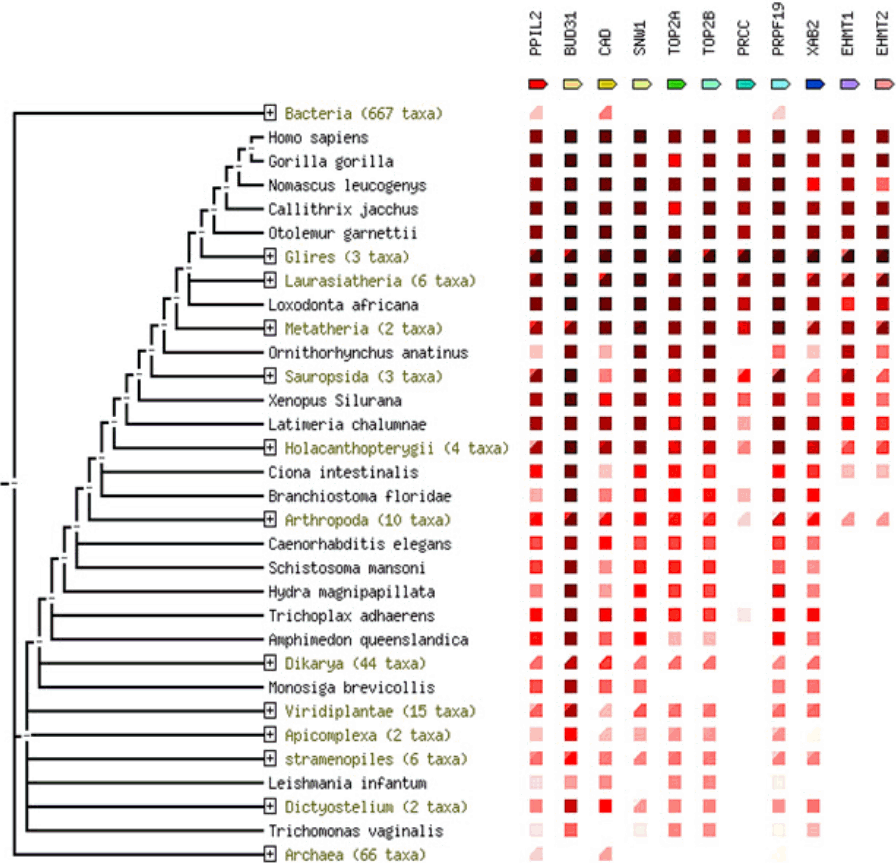
Phylogenetic relationship among PPIL2 gene family in various species by co-occurrence program.
Expression analyses of miR-21-3p and PPIL2 genes in olive flounder tissues
To evaluate the relevance of the expression levels of miR-21-3p and PPIL2, the expression patterns were examined in various olive flounder tissues including spleen, gill, muscle, kidney, intestine, stomach, liver, fin, brain, and testis. The expression pattern of miR-21-3p in multiple tissues was determined by quantitative real-time RT-PCR with miRNA U6 as a reference. The results displayed that miR-21-3p was up-regulated in most of tissues, except for testis. The expression level of miR-21-3p was highest in liver, followed by muscle. Testis expression level was relatively lower than other tissues except liver and muscle tissues (Fig. 8A).
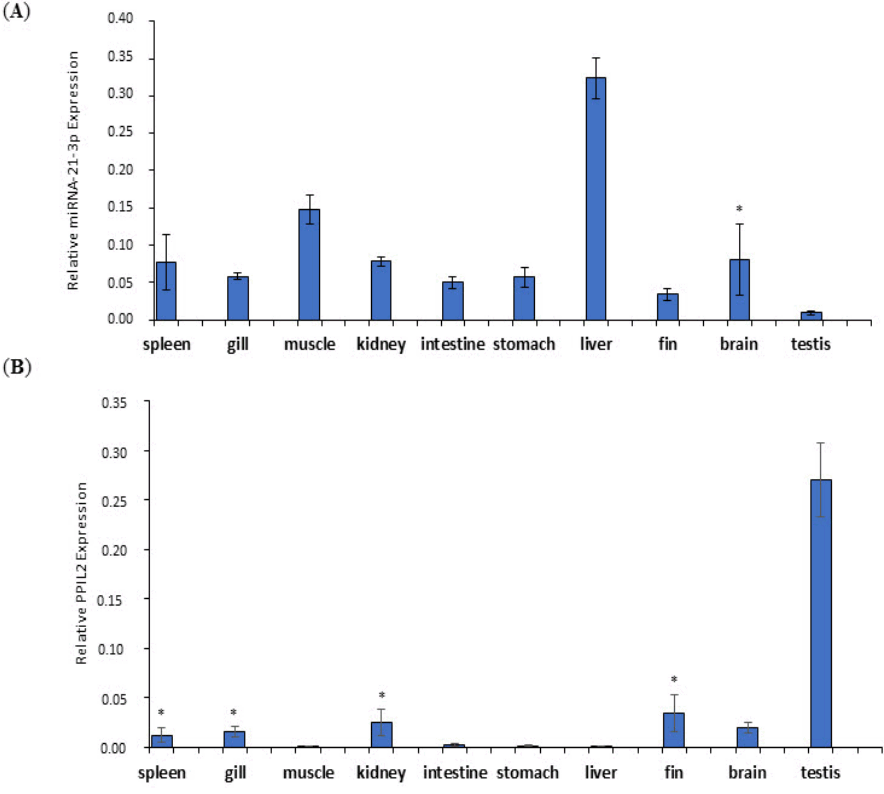
Relative expression patterns of miR-21-3p (A) and PPIL2 (B) in various tissues of olive flounder. Data are normalized to U6 and glyceraldehyde 3-phosphate dehydrogenase in miR-21-3p and PPIL2, respectively. This results are presented as mean ± standard deviation. A paired t-test was indicated to gain p-values and to check statistically important differences in expression levels of miR- 21-3p and PPIL2 (*p ≥ 0.15).
The expression degree for PPIL2 in the numerous tissues were compared with the GAPDH as reference gene. Overall, most of the tissues were down-regulated compared with expression of miR-21-3p. Especially, testis showed the highest expression level of PPIL2 but PPIL2 was hardly expressed in the muscle, intestine, stomach, and liver (Fig. 8B). To sum up, the expression levels of miR-21-3p and PPIL2 mRNA were conversely related to most of tissues.
Discussion
The seed region usually has six nucleotides which affects the most important elements for recognizing and combining 3´ UTR regions [25]. To observe the relationship of evolutionary conservation about these miRNAs, we analyzed those sequences using ECR browser system and genomic approach, indicating that seed region was highly conserved from fish to primates (Figs. 1 and 2). These data allow us to understanding miRNA functional research among those species [26]. In the previously study, miR-21-3p sequence (5´-CAACACCAGUCGAUGGGCUGU-3´) was predicted seed region as 5´-AACACCA-3´ and matched with 7mer of target mRNA 3´ UTR region. Also, miR-21-3p has the potential target gene, FZD4 and it is positively correlated to synergetic reaction about target mRNA silencing activity [27]. In the present study, we found seed region as 6mer 5´-CAGUCG-3´ via miRbase and evolutionary conservation related programs. Then, the sequences were confirmed by using mfold web server (Fig. 3). All target mRNAs of the miR-21-3p by PANTHER program allow us to understand their biological functions. Therefore, we examined GO analysis, showing that most of the target mRNAs seem to be associated with cellular process, cellular metabolic process, catalytic/binding activity, and cell part component (Fig. 4). In the previous study, GO enrichment in the genes noted serious enhancement for target mRNAs. These targets were related to the defense response, inflammatory response, and immunological reactions, which the result revealed nuclear factor-kB signaling pathway by using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway program [28]. Also, the possible regulatory pathways for the miR-21-3p at major target genes were analyzed by KEGG terms. GO categories were related to metabolic process, cell growth, cell cycle process, cell division, and signal transmission [29]. In order to understand the relationship between target gene and miRNAs, target genes were predicted from miRNA target prediction programs [30] and then selected 3´ UTR of the genes which have binding site to seed region of miR-21-3p (Table 1). Since one miRNA can regulate different target genes [25], miR-21-3p can target diverse genes in various species. Among various categories from Table 1, this study focused on MFE score that indicates the degree of affinity between miRNA and its 3´ UTR target region. Within the genes with high MFE score, six genes were selected KANK2, SNAPC3, HN1, AIFM1, RELA, and PPIL2. These candidates for the target gene of miR-21-3p were indicated by multiple bioinformatic analysis. Of these, six target genes were selected and the hybridization between 3´ UTR of the target genes and the seed region of miR-21-3p was demonstrated using the RNA hybrid tool (Fig. 5). One of the highest MFE scores was PPIL2, peptidylprolyl isomerase-like 2 acts as a chaperone with putative ubiquitin ligase activity [31, 32]. Until now, there have been few studies about the function of miR-21-3p, but there are several studies about PPIL2 functions in other fields. PPIL2 related target genes are preserved in primates such as gorilla and gibbon (Fig. 7). Likewise, PRCC encodes proline-rich protein PRCC‒PPIL2 complex, which acts as protein folding, transport and degradation in the brain of Alzheimer’s disease patients in previous study [33]. Other studies have also shown that PPIL2 interacts with intronic-miRNAs such as miR-130b in host genomes [34, 35].
Quantitative expression pattern for the miR-21-3p and PPIL2 genes was examined in various olive flounder tissues in the present study. The miR-21-3p expression level was up-expressed except for testis tissue as shown in Fig. 8A. The expression of miR-21-3p in liver of olive flounder significantly increased compared to the other tissues. This result was previously reported up-regulation for liver tissue of Japanese flounder in miR-17 that involved in Japanese flounder metamorphic developmental stages [16]. As PPIL2 was found to be a target gene of miR-21-3p, this study performed qRT-PCR to confirm the expression patterns of various tissues in olive flounder. The expression levels of PPIL2 gene was highest in the testis, followed by the fin, kidney and brain. This result proposed that PPIL2 has an enzyme activity for peptidyl prolyl isomerase (PPIases), related to function of posttranslational modification that accumulate the isomerization of peptidyl prolyl bonds in peptides and proteins [36]. PPIases can be classified into three families; cyclophilin, FKBP, and parvulins. Among these families, Pin1 belongs to the parvulin family that regulates the function of phosphorylated protein like the cell cycle regulatory molecules [37]. These three families of PPIases activity was up-expressed in wild-type mice testis lysate both mRNA and protein levels [38]. Taken together, our data indicated that PPIL2 gene expression showed complementary results with those of miR-21-3p expression in various tissues of olive flounder.
In summary, genomic and bioinformatics analyses of miR-21-3p allow us to speculate the biological function in relation to the target genes of the miR-21-3p in olive flounder. In various target genes, 3´ UTR region of PPIL2 gene showed the highest binding affinity with miR-21-3p based on the MFE value. Therefore, we examined the expression pattern in both miR-21-3p and PPIL2 gene, indicating that the miR-21-3p seems to repress the expression of PPIL2 gene. Our bioinformatics data could be of great use as further studies in relation to the biological roles of miR-21-3p in various species.
Acknowledgements
This research was provided for a grant the Marine Biotechnology Program (PJT200620, Genome Analysis of Marine Organisms and Development of Functional Application) funded by the Ministry of Oceans and Fisheries, Korea.
Notes
Authors’ contribution
Conceptualization: HSK
Data curation: AJ
Formal analysis: AJ
Methodology: AJ, HEL
Writing - original draft: AJ
Writing - review & editing: AJ, HEL