Introduction
We are in the genome sequence era. Now, we can all read our genome sequences if we have a bit of time and money. We can know our own program of life given by our fathers and mothers, which consists of 3,000,000,000 bp ├Ś 2 of DNA sequence. This knowledge (knowing ourselves) is revolutionizing medicine. The information gives us endless possibilities and a huge number of applications in research and clinical practice of medicine. Preventive medicine, personalized medicine, pharmacogenomics, and pharmacogenetics-these were just dreams on the desk or in the brain of medical scientists and doctors a few years ago but are all achievable at the bedside today.
The best application of the information obtained by genome sequences is an attempt to conquer common diseases or lifestyle-associated diseases. Common diseases are our common problem. It is a serious medical, social, and economical problem all over the world. It's a problem of the present and the future world. The study of our genome has given us a tool to tackle it-the genome-wide association study (GWAS), which is now in full bloom. As we all know, GWASs have succeeded in the identification of susceptibility genes of many common diseases [1].
For more than 15 years, I have been working on genomic studies and committed to the GWAS from its dawn. In this paper, I briefly review a history of human genome research by focusing on GWASs related to my work.
Beginning
Where were you at the start of the new century? What were you doing at that time?
I was in Japan, in Tokyo, in the SNP Research Center (SRC). SRC was founded as a part of the Japanese Millennium Project to study single-nucleotide polymorphisms (SNPs). The Millennium Project is a broad effort to get industry, academia, and government to cooperate on a wide range of R&D projects to revitalize the competitiveness of the industry and to energize Japan's economy. The project focused on developing technologies for three main target areas of vital importance to Japan: informatics, ecology, and an aging society. A variety of large-scale human genome studies were done as projects to revolutionize medicine and cope with the aging society. In April 2000, SRC started collaboration with the Human Genome Center (HGC), Institute of Medical Science (IMS), University of Tokyo, and the Japan Science and Technology Agency (JST). Its mission was to identify up to 150,000 SNPs throughout the human genome within two years, to make the information available to the public, and to develop analytical tools for polymorphisms (note: SRC was renamed Center for Genomic Medicine [CGM] in 2008 [http://www.src.riken.jp/]).
SNPs are the most common form of DNA sequence variation. They are the most useful and convenient polymorphic markers to map genes that modify disease susceptibility or those related to drug responsiveness. If we had a dense catalog of SNPs, we could narrow down the loci relating common disease susceptibilities to a small region containing one gene or so by linkage disequilibrium mapping. To get such a catalog, the discovery of a large number of SNPs was first necessary [2].
Draft Sequence to the First GWAS
The draft sequence was published in 2001 [3]. It was a collaborative work of the public sector via the international Human Genome Project (HGP) and the private sector via Celera Genomics. HGP refers to the international 13-year effort, formally begun in October 1990, to discover all the human genes and make them accessible for further biological study. Another project goal was to determine the complete sequence of the human genome. Actually, the latter group led by Dr. Craig Venter, was the critical player for the success of finishing the sequence, which invented the strategy of the shotgun sequence, followed by computational assembly [4]. It was a revolution in genomic research.
With the help of the draft sequence, Japanese Single-Nucleotide Polymorphisms (JSNP), the first population-specific SNP database (http://snp.ims.u-tokyo.ac.jp/), was established as a collaborative work of HGC-IMS and JST [5]. A total of 190,562 genetic variations (mainly SNP) in 24 subjects were discovered and confirmed and have been made available through this website [6].
Based on information in the JSNP database, Tanaka's group in SRC succeeded in identifying a susceptibility gene for myocardial infarction [7]. This study was the first GWAS in the world [8, 9]. By a two-stage association study-i.e., a GWAS followed by replication of a limited number of the SNPs with significant p-values-they found a functional SNP in the lymphotoxin-╬▒ (LTA) gene associated with myocardial infarction. Their GWAS was quite primitive by current standards. They used only ~100,000 SNPs from the JSNP database, because in those days, the number of genes was considered to be ~100,000, and basically, one gene is expected to be covered by one SNP. Both of these assumptions have been subsequently disproven by the HGP [10] and the HapMap Project [11]. The coverage of the genome in the GWAS was low, because the success rate for this genotyping was only 71%. The estimated power (╬▓ value) for detection of a locus was also low, because the number of subjects was very small. However, the basic idea for the current GWAS, including linkage disequilibrium mapping for identifying the most associated SNPs and a pooled control strategy, were already present in the frontier study. Their study clearly showed that the GWAS was a reality, not the dream of statistical geneticists, and that a denser map of common SNPs was indispensable for realizing the dream.
HapMap
Completion of human genome sequencing was announced on April 2003. Reading of 99% of the gene-containing part of the human sequence was finished to 99.99% accuracy [10]. However, in a true sense, the human genome sequence had (and has) not been finished. There are still many gaps in the "reference" sequence-for example, in the centromeres-and given the variation of the human genome sequence, many must wonder what the completion of sequencing the human genome is in the first place.
After completion of the sequencing of the human genome, our center, SRC, joined The International HapMap Project from its start. The project was launched in October 2002 to create a public, genome-wide database of common human sequence variations, providing information needed as a guide to genetic studies of clinical phenotypes [2]. The HapMap Project is a natural extension of the HGP. While the reference sequence of the genome constructed by the HGP is informative about the vast majority of bases that are invariant across individuals, HapMap focuses on DNA sequence differences among individuals. The main genome researchers in the world got together and made an infrastructure of the association study-i.e., a database of 1 million genetic variations for 4 representative populations: Africans, European Caucasian, Chinese, and Japanese. The Africans were the Yoruba people in Ibadan, Nigeria. The European Caucasians were a Utah, USA population with Northern and Western European ancestry collected in 1980 by the Centre d'Etude du Polymorphisme Humain (CEPH). The Chinese consisted of unrelated Han Chinese in Beijing, China. The Japanese were unrelated self-identified Japanese living in Tokyo, Japan. In the HapMap Project, we are proud to say that SRC made the World No. 1 contribution as a research group by genotyping ~1/4 of the genome (Table 1).
HapMap data document the generality of hotspots of recombination, a block-like structure of linkage disequilibrium and low haplotype diversity, leading to substantial correlations of SNPs with many of their neighbors [11]. There is the extensive redundancy among nearby SNPs, providing the potential to extract extensive information about genomic variation without complete re-sequencing, as well as efficiencies through selection of tag SNPs and optimized association analyses.
Gold Rush
After HapMap, GWAS became as easy as expected and intended; to perform a GWAS, we just collect DNA from patients, genotype it by using commercial arrays, and do a bit of statistics for the results (actually, in most cases, commercial companies do it for us as a business) (Fig. 1). Genotyping has become work, not research; commercial genotyping systems have spread all over the world. By sharing the controls, collecting controls is no longer necessary, except for diseases with extraordinary high prevalence (>10%). A huge number of susceptibility genes are mapped by this 'easy' method. As a consequence, after HapMap, as commercial genotyping systems (Illumina, etc.) have spread, the association study has spread all over the world.
As a result, a gold rush has come [1]. The study of genetic polymorphisms (SNP, copy number variation, etc.) with its application to disease studies was selected for the No. 1 scientific event in the year 2007 [12]. Naturally, the application refers to the identification of susceptibility genes for common diseases by GWAS. You must have seen (and are still seeing) so many GWAS papers in good journals, which have reported susceptibility genes for so many common diseases, including obesity, hypertension, diabetes, and osteoporosis. For example, Nature Genetics is one of the top journals in the field of genetics, with an impact factor in 2010 of 36.377, and in that year, it published a total of 181 papers, of which 56% was GWAS-related. So, for the journal, the genetic study is almost equal to the association study.
There came such a big wave of medical science, and of course, we got on the wave. We did so many GWASs that were the first for diseases, including GWASs for osteoarthritis [13], rheumatoid arthritis [14], and diabetic nephropathy [15]. The big wave has also spread to East Asia. Many good GWAS papers have been published from Korea and China as fruit of this international collaboration [16-18].
End of Beginning: Common Problems of GWASs
Good days are ending, however. Today, the initial enthusiasm for GWASs is decreasing, and the GWAS society is facing severe criticisms. A frequent complaint is that GWAS results mean little to patients due to the small effect of variants on disease risk and their relatively small contribution to common disease etiology. This claim might be partially correct. In the first place, "significantly associated" does not mean that the association identified by the study is significant in biology, medicine, or actual life. For example, a paper reported the identification of a SNP that is associated with height, but its estimated additive effect is only 0.44 cm on height [19]. Another example of dubious significance is a GWAS study claiming that cartilage thickness of the hip joint measured on X-ray films is decreased in subjects with a susceptibility allele; they have a 0.07-mm (not cm!) narrower joint space than those who do not have the allele [20].
However, the low risk (low odds ratio) is not always due to the GWAS itself. In many studies, it is the problem of particular studies themselves, not of the GWAS. For examples, using pooled controls of general populations as controls of highly prevalent diseases, like diabetes mellitus, hypertension, and obesity, must decrease the odds ratio of the susceptibility alleles because of contamination of the patients in the controls. Also, ambiguous definition of populations, like "UK Caucasians" or "Western Europeans," would compromise the result by admixture of different ethnic subgroups. Thus, it is necessary to detect population stratification prior to association analysis.
Moreover, a GWAS is not a method to conclude something. It's a method to map the gene: an associated variant is not necessarily a true causal variant. GWASs present candidates of the causal variant. To identify and prove the causality, we need to find the best associated variations by fine mapping around the smoking gun GWAS signal, followed by functional studies for them. Statistics must lead to biology. Biology must lead to medicine. It is true that after all, no treatment, no benefit, to the patients has been given for common diseases from GWAS results so far. My comment is not intended to look down on GWASs. It is a method to start something; it gives us wonderful start points. You can start the work based on the reality of the disease-clinical evidence (patient, clinical record) from the human evidence in front of you-to the patients at your bedside.
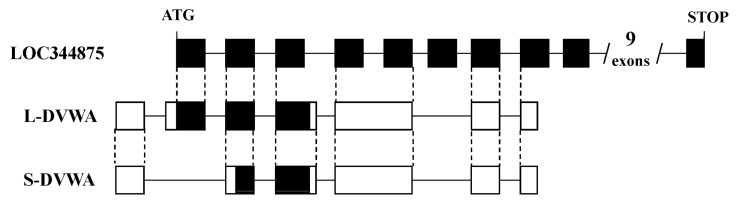
Another problem is that not infrequently, the interpretation of the 'mapping' results is wrong. In many studies, authors just make a story for causality by relating to known genes near the marker SNPs, even if they are far apart on the genome. The association does not tell which gene is implicated; it just tells which sequence variation is wrong. Some of the readers might be thinking that all genes have already been discovered by the international genome project. This is totally wrong. There are still many more genes that have not been identified. For example, we have found a novel susceptibility gene, DVWA, that is associated with osteoarthritis [21]. DVWA was in a gene desert region when we first found it in 2004. While we were working on cloning it [21, 22], a computer-predicted gene was mapped to the region; however, the prediction was a mess. The number of coding exons was predicted to be 19, while it was actually 3. Only 1 in 7 experimentally determined exons of DVWA had been correctly predicted regarding the exon-intron boundary and coding region (Fig. 2). We also found a novel susceptibility gene for osteoporosis, FONG [23], which was not predicted correctly in the public database. GWASs can be a starting point to find a hidden gene in the genome, and we should remember that now, the concept of the "gene" itself is changing. We have already known that there are so many non-protein-coding genes: the genes encoding functional RNAs [24]. They are quite compatible to the original concept of the gene: information in the genome that determines phenotype or trait. To code proteins is just one of the many ways to determine phenotype.
Still Other Problems of GWASs: Lesson Learned for the Future
GWASs have so many false positives. p < 10-2 (1% significance level) means a 1 in 100 coincidence. If you try 1,000 times, you can hit 10 targets and 100,000 times, 1,000 targets-all quite by chance. Therefore, association needs replication, but repeating in the same population is not a good idea; it might just introduce the same error in the first study, which is sometimes difficult to detect. Studies in different populations, preferably in populations from different ethnic groups, are desirable. Because one population is one experiment of nature, one ethnic group is one experiment given from heaven. Every experiment is precious. Multiple replications in large samples provide the most straightforward path for identifying robust and broadly relevant associations. International collaboration is necessary. For international replication, East Asia has a definite advantage when we consider its relative genetic similarity among the subpopulations, large population size, and high medical standard of the countries in the area.
Real Value of GWASs
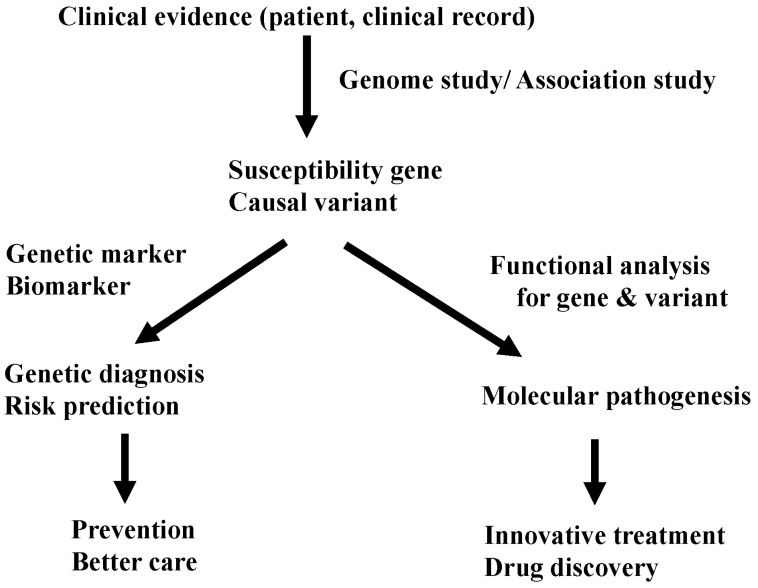
Currently, most people look at GWASs as a method to identify a marker for genetic diagnosis and risk assessment. Yes, by knowing the genetic risk, we can re-evaluate and modify our lifestyle and prevent unhappy problems, like side effects of drugs. However, GWASs could give us far sweeter fruit. Through functional analysis of a gene and its associated variants, we can approach the mechanism of the disease. By studying the meaning of allelic differences of causal variants, we can clarify the molecular pathogenesis of the disease. With the knowledge of the pathogenesis, we can walk directly to develop innovative treatments and discover new drugs (Fig. 3). Therefore, identification of the significantly associated SNP is just 'end of the beginning' to get to the final goal: treatment of patients with common diseases. However, at present, after identification of 'marker' SNPs by GWASs, we have no golden road to find 'causal' SNPs (variants). We have no customized method to identify functional variants. There is still a long and winding road before us. It's just the beginning.