SFannotation: A Simple and Fast Protein Function Annotation System
Article information
Abstract
Owing to the generation of vast amounts of sequencing data by using cost-effective, high-throughput sequencing technologies with improved computational approaches, many putative proteins have been discovered after assembly and structural annotation. Putative proteins are typically annotated using a functional annotation system that uses extant databases, but the expansive size of these databases often causes a bottleneck for rapid functional annotation. We developed SFannotation, a simple and fast functional annotation system that rapidly annotates putative proteins against four extant databases, Swiss-Prot, TIGRFAMs, Pfam, and the non-redundant sequence database, by using a best-hit approach with BLASTP and HMMSEARCH.
Introduction
Functional annotation of putative proteins is a fundamental and essential practice in the postgenomics era [1]; it allows us to analyze genomic and genetic features, such as physiological activity and metabolism, as well as to discover medically and industrially relevant enzymes. Since large numbers of putative proteins were discovered from a vast amount of sequencing data generated using high-throughput sequencing technologies, including those of the next and third generation, many automated functional annotation systems have contributed greatly to the annotation of them with minimal manual effort [2]. However, their runtime performance of functional annotation against large extant databases often causes a bottleneck, and especially, standalone tools, such as AutoFACT [3] and BLANNOTOR [4], demand high-performance hardware resources for fast annotation from users.
From the user's perspective, a web-based annotation server system would be a useful tool to bypass the demands of high-performance computer resources, and besides, they offer user-friendly interfaces. The RAST server system is particularly popular and can be used to rapidly annotate many microbial proteins against a specially curated subsystem database [5]. Web server systems, however, may be undesirable because of critical obstacles, such as the limitation of usable server resources, a long waiting time by many queries, a low-bandwidth network or unstable traffic flow associated with the upload of query data and download of outputs, and data security problems. Thus, some users prefer standalone systems to web-based systems in spite of the demand for high-performance resources. Although standalone and web-based systems have good and bad points, slow runtime performance in themselves cannot be avoided because of the exponential increase in database sizes, without controlling some aspect of the annotation workflow.
We developed SFannotation, which rapidly annotates putative proteins by using single or bidirectional best-hit approach with sequence-based methods-BLASTP [6] and HMMSEARCH [7]-against big extant databases: Swiss-Prot [8], TIGRFAMs [9], Pfam [10], and the non-redundant sequence database (NR) of NCBI [11]. As best-hit approaches, especially bidirectional best-hit [12], have been widely utilized in searching reliable homologous protein sequences, such as orthologs, as well as functional annotation systems [13,14,15,16], SFannotation can reliably annotate putative proteins. Remarkably, SFannotation can rapidly annotate proteins against large extant databases by our hierarchical workflow.
Methods and Results
Before annotating putative proteins against Swiss-Prot, TIGRFAMs, Pfam, and the NR database, SFannotation filters out all proteins described in the databases by terms, such as "unknown," "hypothetical," "unclassified," "uncharacterized," "putative," "predicted," and "conserved" (Fig. 1A), because some putative proteins may be misannotated by their inclusion. Then, using BLASTP and HMMSEARCH, SFannotation searches homologous proteins and domains in each refined database using a default threshold (≤10-5 E-value) and selects the highest-scoring homolog to annotate putative proteins as the best-hit approach, such as single best hit and bidirectional best hit [12, 16].

Database filtration (A) and workflow of the SFannotation annotation system (B). Black arrows represent putative proteins that are annotated by the best-hit approach, and red arrows represent the conversion of unannotated proteins to query putative proteins to search homologs against other databases.
Putative proteins are hierarchically annotated using the following database priority: Swiss-Prot → TIGRFAMs → Pfam → NR, which is ordered according to their reliability (Fig. 1B). Once annotated, the putative proteins are no longer queried using homology searches against the other databases. For example, if a putative protein is annotated against Swiss-Prot, it is excluded from annotation against the other databases, while the remaining unannotated putative proteins continue to be annotated against the other databases. Therefore, the runtime performance can be reduced, because the number of unannotated putative proteins gradually decreases (Fig. 2).
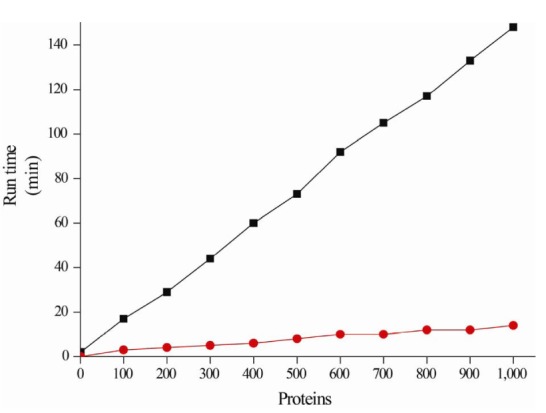
Runtime of the SFannotation system (red) and a best-hit approach without the hierarchical SFannotation workflow (black). Randomly selected proteins from Escherichia coli MG 1655 (GenBank accession number: U00096) were tested using a 64-bit Linux system (Ubuntu) possessing 20 CPU threads.
Implementation
SFannotation is written in Perl and bash shell and is implemented on a Linux/Unix system on which BLASTP and HMMSEARCH are able to function. SFannotation automatically annotates putative proteins with downloading of all four databases, as well as BLASTP and HMMSEARCH. SFannotation is implemented by a command line on the Linux/Unix system: "perl SFannotation --download --fasta <input fasta file> --speedup" (Supplementary Fig. 1).
Acknowledgments
We thank the members of the Korean BioInformation Center (KOBIC). This project was supported by a grant from "KRIBB Research Initiative Program" and the Korean Ministry of Science, ICT & Future Planning (MSIP) under grant numbers NRF-2010-0029345 and NRF-2011-0019745.
Notes
Availability: SFannotation system is available at https://code.google.com/p/axxa76/wiki/SFannotation.
References
Supplementary material
Supplementary data including one figure can be found with this article online at http://www.genominfo.org/src/sm/gni-12-76-s001.pdf.
Supplementary Fig. 1
Snapshot for the various options and execution of SFannotation on Linux.