Introduction
Since large-scale epigenetic datasets such as Encyclopedia of DNA Elements (ENCODE) or Roadmap Genomics became publicly available [1, 2], there has been a growing interest in predicting the function of non-coding DNA regions directly from their sequences [3ŌĆō6]. The details of the dynamics of chromatin state conversions among different cell types reported that extensive signal variation exists in regulatory regions [7]. And recent studies based on ChromHMM datasets [8, 9] provided novel insights into n-gram probabilistic language models for non-coding DNA regions stratified by chromatin variability.
As a follow-up to our preliminary study on ChromHMM datasets of ENCODE [10], we extend our discoveries and continue ongoing efforts to build comparative nucleotide frequency profiles stratified by the chromatin variability. We hope to detect Markov properties by analyzing datasets of the full range of 127 cell and tissue types provided by Roadmap Genomics.
We investigated whether some subsets of the annotated Roadmap Genomics 15-state model stratified by chromatin variability can be predicted by purely making n-gram models of DNA sequences. To do that, ChromHMM blocks of human genome were first dissected into nucleosome resolution of 200-bp units, which accounted for 1,965,764,166 units (127 ├Ś 15,478,458 of 200-bp units), and they were integrated into one BED file. Then each individual unit was assigned one dominant chromatin state, by analyzing the integrated BED file of ChromHMM. Next based on chromatin variability of each 200-bp unitŌĆöreferred to as occurrence frequencyŌĆöthe number of different chromatin states in ChromHMM annotations we divided the units into two groups for each chromatin state: highly variable units and invariable units. By using highly variable 200-bp units as our control group, we were able to isolate some invariable chromatin units that showed strong Markov properties.
Methods
The process is explained in detail in the following sections and the basic steps include: combining 127 BED files into a single file, sorting 200-bp units by the frequency of chromatin variability, filtering out highly variable 200-bp units, building 5th order Markov Models, and evaluating prediction accuracy.
Combining 127 BED files into a single file
The Roadmap Epigenomics consortium has released a 15-state model of BED files from a joint analysis of 111 consolidated epigenomes with 16 additional epigenomes from the ENCODE project [2] in total 127 BED files for public download [11]. We downloaded 125 ChromHMM BED files from the Roadmap Genomics (E001.bed through E128.bed files). We excluded E60.bed and E64.bed files, as of August 20, 2017, to build comparative nucleotide frequency profiles (with human genome GRCh35/hg19) to detect their Markov properties.
The 15-state ChromHMM model consisted of eight active states and seven repressed states: active transcription start site (TSS), proximal promoter states (TssA, TssAFlnk), a transcribed state showing both promoter and enhancer signatures (TxFlnk), actively transcribed states (Tx, TxWk), enhancer states (Enh, EnhG), zinc finger protein genes (ZNF/Rpts), heterochromatin (Het), bivalent regulatory states (TssBiv, BivFlnk, EnhBiv), repressed Polycomb states (ReprPC, ReprPCWk), and a quiescent state (Quies) [2].
When making 15-state ChromHMM BED files, the Roadmap Genomics consortium used a core set of five chromatin markers [2]. We investigated whether some subset of the annotated Roadmap Genomics 15-state model can be predicted by making pure n-gram models of DNA sequences, in reverse. To do that, ChromHMM blocks of human genome were initially dissected into different nucleosome resolution of 200-bp units and each individual unit was assigned one dominant chromatin state, by analyzing the 127 BED files of ChromHMM.
To elaborate on our method, BED format shown at the top of Fig. 1 shows a flexible way to define the data lines that are displayed in an annotation track. The four BED fields shown in each of the BED file represent chrom (the name of the chromosome), chromStart (the starting position of the feature in the chromosome), chromEnd (the ending position of the feature in the chromosome), and state (the 15 chromatin states of Roadmap Genomics, ranging from 1 to 15). For example, the chromatin state of E001 in Fig. 1, for the block from chr1: 9,800 to chr1: 10,600 is 9 (Het heterochromatin state), whereas the chromatin state of E002 in Fig. 1, for the block from chr1: 762,000 to chr1: 763,000 is 1 (TssA proximal promoter state).
For our study, it became critical to develop a functional annotation framework that could be generalized to different cell types. To build good predictive models in making the Markov models of human genomes, we modified the original BED files by dissecting ChromHMM blocks in each BED file into 200-bp units. For example, the original unit of E001 cell line in Fig. 1, ranging from chr1: 9,800 to chr1: 10,600 (a unit size of 800-bp) was dissected into four units of 200-bp blocks (from chr1: 9,800 to chr1: 10,000; from chr1: 10,000 to chr1: 10,200; from chr1: 10,200 to chr1: 10,400; and from chr1: 10,400 to chr1: 10,600), in a new BED file. Likewise, the original E002 unit in Fig. 1, ranging from chr1: 762,000 to chr1: 763,000 (a unit size of 1,000-bp) was dissected into five units of 200-bp units. Profiling nucleotide frequency tables by units of 200-bp is a convenient way to build a general framework and test various Markov properties simply by joining these 200-bp frequency tables differently for specific outcomes and resolutions.
By dissecting the units uniformly, it became possible to combine all the annotations spread out through 127 different BED files, into a single integrated BED file, as shown in the bottom of Fig. 1. Each row of the integrated BED file is composed of eighteen entries from the original BED files: chromosome number, unit starting number, unit ending number, and the number of annotation occurrences of each of fifteen chromatin states. For example, chr1: 12,800ŌĆō13,000 unit in bottom of Fig. 1 shows that this specific 200-bp unit is annotated 28 times as state 5 (TxWK), 4 times as state 7 (Enh), and 94 times as state 15 (Quies) throughout the original 127 BED files, whereas occurrence count numbers of all remaining chromatin states for this unit are zero, for the 1, 2, 3, 4, 6, 8, 9, 10, 11, 12, 13, and 14 states.
Filtering out highly variable 200-bp units
Since the details of the dynamics of chromatin state conversions among different cell types was reported it was noted that extensive signal variation exists in regulatory regions [7]. So, we needed a way to quantify signal variation in regulatory regions. Thus, we defined the unit variability count of chromatin states of a given 200-bp unit as the number of states where counts of occurrences were non-zero, to define and compare the observed consistency of each chromatin state at any given genomic position across all 127 epigenomes.
Table 1 shows some randomly chosen highly variable and invariable 200-bp units of the integrated BED file, sorted by chromosome number. The degree of chromatin variability is marked as ŌĆśHŌĆÖ (high) or ŌĆśLŌĆÖ (low), as in the last column of the table. According to the table, the chromatin state variability count of unit chr14: 61,114,600ŌĆÆ61,114,800 unit would be eleven, as there were eleven non-zero states (state 1, 2, 3, 5, 7, 10, 11, 12, 13, 14, and 15), and this unit is marked as ŌĆśH,ŌĆÖ or highly variable.
We reasoned that we could use variability count statistics and the maximum likelihood decision rule to make optimal classifications for the Markov model, since uniform priors can be assumed if we only use 200-bp units with low chromatin state variability. In this way, highly variable 200-bp units where different chromatin states were frequently switched to other states across different tissues and cell types could be either eliminated, or used as controls in datasets, in practicing Markov models.
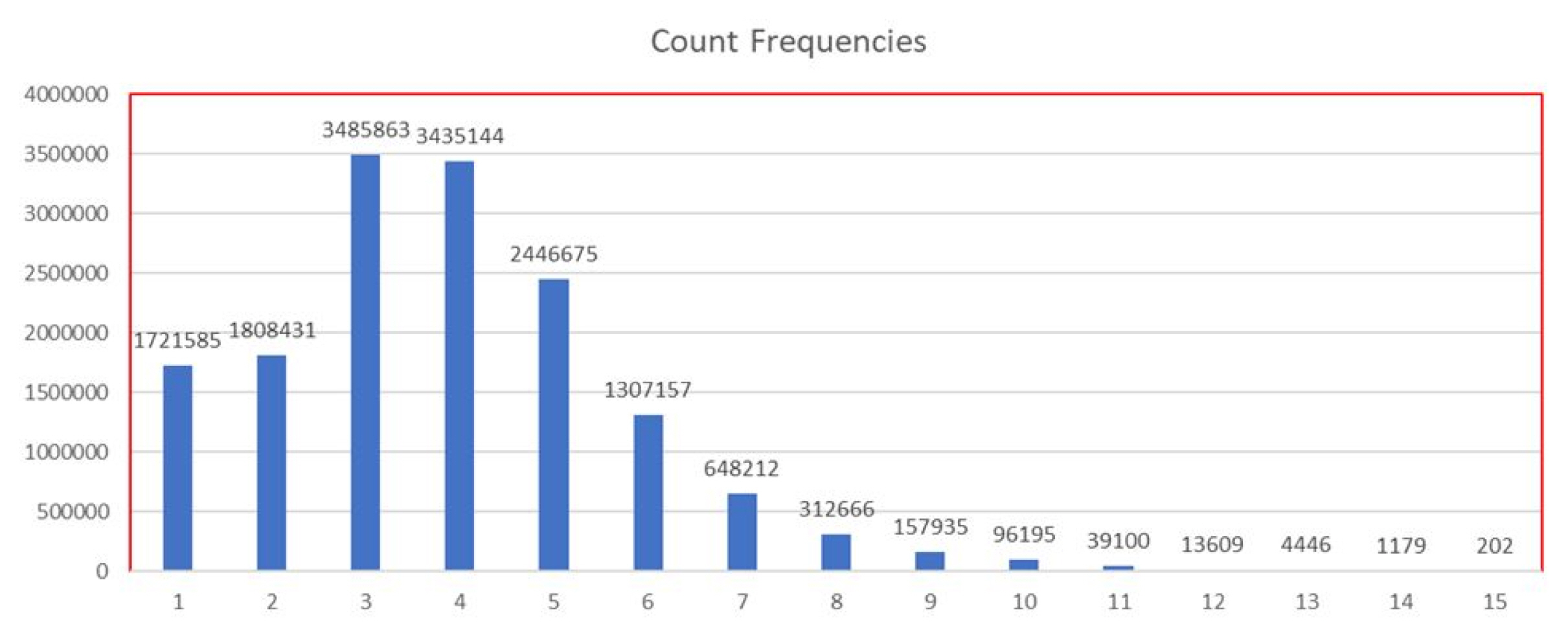
Fig. 2 shows statistical distribution of variability counts across human genomes. Human genomes are dissected into 200-bp units from the original 15,478,458 units. Among them, the variability counts of 1,721,585 units were one, meaning that all 127 cell lines annotated the same state in these 200-bp units. The variability count of chromatin states of 1,808,431 units was two; meaning that all 127 cell lines were annotated as one of two states in these 200-bp units.
Furthermore, if a state with the number of occurrence count fewer than five was discarded in each of the units, the average variability count of chromatin states drops dramatically. Variability counts of less than 3 states accounted for 93.64% (38.59% + 36.87% + 18.18%) of overall 200-bp units. This means that most of these 200-bp units have a strong preference for a certain chromatin state.
Our model should not be based on a single cell line, therefore; it is critical to propose a new functional annotation framework that can be generalized to different cell types. This gave us good heuristic insight to design new Markov models for our study. A generalizable framework can be achieved through statistically-justifiable models. Based on the newly integrated BED file, we assigned a dominant chromatin state for each of the 200-bp unit, as was explained in the previous section. In this way, it was possible to assign just one or two dominant chromatin states for most of the 200bp units of the entire human genome.
Building two-state fifth order Markov models for each of the 15 chromatin states
A Hidden Markov model (HMM) is a probabilistic model. The key property of a Markov chain is that the probability of each symbol xi depends only on the value of the preceding symbol xiŌłÆ1 [i.e., P(xi |xiŌłÆ1)], not on the entire previous sequence [i.e., P(xi |xiŌłÆ1, ŌĆ” , x1)].
Usually, it consists of states corresponding to a biological meaning (e.g., chromatin states) and allows transitions between these states in a biologically meaningful way. The model can define a probability distribution on DNA sequences together with chromatin states.
Our rationale for using HMMs was that invariable units contained in ChromHMM chromatin states can follow a regulatory grammar. They do not form a short fragment of motifs or DNA signatures, but form as a continuous, longer stretch of sequences.
Thus, the 200-bp units were subdivided into 15 chromatin states, based on dominant states. Then, based on chromatin variability of each 200-bp unit, we further divided the 200-bp units into two groups for each of the chromatin state: highly variable units and invariable units.
Fig. 3 visualizes our approach. After we assigned a dominant chromatin state for each 200-bp unit, the integrated BED file was sorted according to chromatin variability count and frequency counts, for each of the 15 chromatin states. Then, for each of the fifteen chromatin statesŌĆÖ top 1,000 units (ŌĆśLŌĆÖ or invariable 200-bp units) and bottom 1,000 units (ŌĆśHŌĆÖ or highly variable units) were selected for each of the 24 chromosomes. By trial and error, we rebuilt newer Markov chains by iteratively analyzing the variability count of chromatin states of a given 200-bp unit. Samples were stratified by chromosomes into strictly non-overlapping training, and testing sets. A total of 720,000 200-bp units were trained were used for training HMM models:
And an additional 72,000 200-bp units were tested for prediction accuracy. Mostly, we profiled each 200-bp with chromatin states and built new transition tables by training the 200-bp blocks with chromatin variability less than 2, if possible.
Results
We directly investigated whether our HMMs based on invariable units have the discriminating power against control datasets of highly variable units. As these HMMs could be used as a binary Naive Bayes classifier, we calculated the sequence of each 200-bp unit that maximized our Markov models. For each chromatin state, the given HMM tries to capture the statistical differences in the two hidden states of ŌĆśHŌĆÖ (High) and ŌĆśLŌĆÖ (low). As was shown in Fig. 3; based on nucleotide frequency profiles and given a random sequence x1,x2, ŌĆ” ,xn, we calculated sequences ŽĆ1, ŽĆ2, ŌĆ” ,ŽĆn of chromatin states that maximized the probability between highly-variable and invariable states, for the 15 ├Ś 2 Hidden Markov models.
Table 2 shows the prediction accuracy for the 15 chromatin states. We defined a correctly predicted unit as one when predicted results matched the dominant chromatin state of annotations of each of the testing units against control datasets or highly-variable units.
According to Table 2, HMM trained from invariable 1_TssA-dominant units shows the highest prediction accuracy of 85.3%. Investigating the dynamics of chromatin state conversions of highly variable units, we found that most significant state switches are between active states. Among eight active chromatin states, the order of prediction accuracy can be sorted as follows.
Among seven inactive chromatin states, 10_TssBiv and 11_BivFlnk, and 12_EnhBiv showed a high precision rate above 90%.
These results and additional properties of the model suggest that n-grams related to invariant chromatin regions are an inherent and biologically-informative feature of the genome. The framework enables us to infer about coordinated differences in marks by studying chromatin state variability of 200-bp units.
Discussion
We extended our previous study of conditional characterization of the Markov property of publicly available chromatin states by building new Markov models of the active chromatin states.
Table 3 shows average occurrence frequencies of a dominant state of invariant units for the 15 chromatin states. In doing so, we found that some inactive chromatin states were highly constitutive and marked in most of the 127 epigenomes. For example, state 15 (Quiescent state) covered on average 68% of each reference epigenome. Thus, the occurrence frequencies of 15_Quies state for the top 24,000 units were all 124.0 (e.g., chr Y: 13,107,200 to chr1: 13,107,400 units in the bottom of Table 1). This explains the reason why recall of state 15 was relatively high (91.0% in Table 2). The average frequencies for the state three and six were 20.0 and 36.7, respectively. These states are not usually in a dominant state in most of the 200-bp units, and thus are not appropriate for HMM models.
One limitation of our study is that when building HMM models from 200-bp units of DNA sequences, we did not consider whether the unit should be read forward or in reverse, at this time. We just averaged the n-gram counts of both cases when building emission or transition tables.
Another limitation of our study is that the unit length is set at 200 base pairs without exception. The states of HMMs models based on chromatin states can have an explicit length distribution of the sequence emitted in chromatin state. Future HMM models should consider the DNA sequences of states together with emission lengths. Due to the current lack of data availability this is beyond the scope of this paper.
In conclusion, identifying functional regions in the human genome is a major goal in human genetics. N-gram models, including the most notable n-gram model the Hidden Markov model, have been extensively studied in the field of bioinformatics. However, the Markov property of nucleotide sequences associated with chromatin states at whole human genome scale has rarely been reported in the literature. Consequently, little research has been carried out to explore n-gram models associated with whole genome-wide chromatin maps.
It is important to note that we only used DNA sequences contained in epigenetic datasets in modelling Markov chains. Our study showed that some subsets of the invariable chromatin states possessed strong Markov properties. Though our study is preliminary, it is significant in that it has potential to be used to construct statistical models necessary for developing algorithms to predict function directly from sequence when combined with SNPs, motifs, and other resources in future studies.