Introduction
Non-coding RNAs (ncRNAs) are defined as RNA transcripts that are not translated into proteins. Classically, only a limited number of RNAs, including ribosomal RNAs, transfer RNAs, small nuclear RNAs, and small nucleolar RNAs, were perceived as ncRNAs. However, this view changed completely after in-depth analysis of mammalian transcriptomes (reviewed in Mattick and Makunin [1]). A decade ago, the advent of next generation sequencing and high-resolution genome tiling arrays revealed that up to 70% of the human genome is transcribed but only about 2% is occupied by protein-coding regions. Moreover, some ncRNAs were found to play regulatory roles for gene expression, as compared to classical ncRNAs that play housekeeping roles. So, ncRNAs are much more dominant in quantity and more diverse in function, than they were thought before.
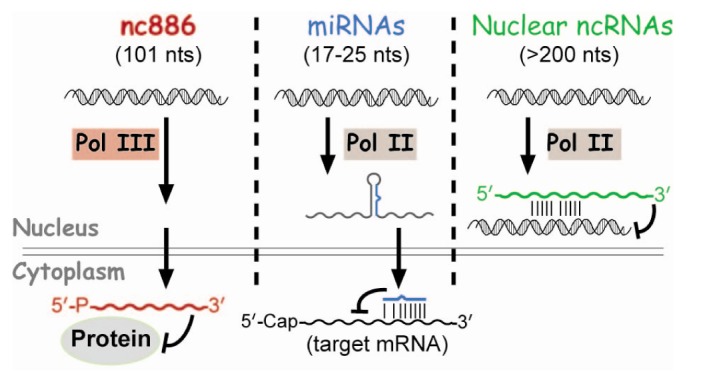
These regulatory ncRNAs have been a topic of intense study during the last decade. So far, most regulatory ncRNAs bind to target genes via nucleic acid interaction to serve as a specificity factor to recruit effector proteins (Fig. 1). For example, nuclear ncRNAs recruit chromatin remodeling factors and microRNAs (miRNAs) recruit the mRNA-slicing complex (reviewed in Fatica and Bozzoni [2] and Filipowicz et al. [3]). Our laboratory has recently discovered a ncRNA that functions in a completely different way and we named it nc886 [4,5]. nc886's function is to bind to a protein and control its activity, rather than to recognize target DNA or RNA. Here, a summary of its characteristics and roles in cancer will be reviewed.
nc886: Identity
nc886 has been given several names, including vtRNA2-1, CBL3, and pre-miR-886. vtRNA2-1 is currently its official name and stands for vault RNA 2-1 [6]. The vault RNAs (vtRNAs) are the RNA components in the vault complex, a large ribonucleoprotein particle whose precise function has not been elucidated yet (reviewed in Berger et al. [7]). In the human genome, there are three canonical vtRNA genes, vtRNA1-1, 1-2, and 1-3. nc886 was thought to be a vtRNA paralog because of sequence homology and therefore was named vtRNA2-1 [6]. However, nc886 is less similar in sequence to canonical vtRNAs than they are similar to each other [5]. Furthermore, nc886 is neither in physical association with the vault complex nor necessary for the integrity of the complex [5]. nc886 is functionally distinct from vtRNA1-1 [4,5,8,9,10].
Another alias for nc886, CBL3, stands for cord blood lymphocyte (CBL)-derived ncRNA 3. This name originated from the fact that nc886 was one of the most induced ncRNAs when CBL was infected with the Epstein-Barr virus (EBV) [11]. Interestingly, canonical vtRNAs were also induced by EBV, which also contributed to the early misconception of nc886 as a vtRNA [12].
In addition, nc886 was once classified as a miRNA precursor and so was registered in the miRNA database (miRBase: http://www.mirbase.org/) [13]. This was due to the high sensitivity of the next-generation sequencing technologies, together with the high intracellular expression level of nc886 but its short half-life [5]. Thus, its degradation products are continuously produced at a quantity high enough to be captured by small RNA sequencing. According to the deepBase (http://deepbase.sysu.edu.cn/) where small RNA sequencing data were collected and displayed [14], the nc886 sequence was captured at significantly higher frequency than most other genomic regions. In addition, 20-30 nucleotides at the 5'- and 3'-end of nc886 were captured more prominently than its middle portion. This sequencing pattern misinterpreted as each end corresponding to mature miR-886-5p and -3p respectively. However, our extensive analysis proved that it is not a miRNA precursor. First and foremost, nc886 appears as a single band at 101 nucleotides (nts) in Northern hybridization [4,5,8,9,10]. Second, the biogenesis of nc886 is dissimilar to that of miRNAs; nc886 is neither processed from Drosha cleavage nor diced into mature miRNAs by Dicer [5]. Third, there is no inverse correlation in expression between nc886 and the predicted target genes of miR-886-5p or -3p, which is against our expectation if nc886 would to produce functional mature miRNAs [5].
Since nc886 does not produce a miRNA precursor nor is in the vault complex, its aliases pre-miR-886 and vtRNA2-1 are misleading. Therefore, our research group renamed it non-coding 886 (nc886) [4] and this name will be used throughout the review.
nc886: Transcription, Expression, and Genomic Locus
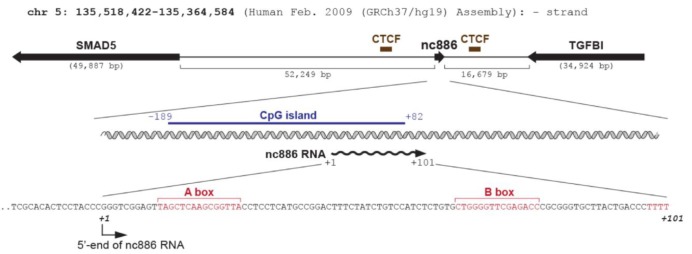
Whereas many other regulatory ncRNAs are transcribed by RNA polymerase II (Pol II) (Fig. 1), nc886 is transcribed by RNA polymerase III (Pol III) as evidenced by its sequence and experimental data. nc886 contains the well-conserved Pol III promoter elements Box A and B followed by an oligo-dT sequence, which is a Pol III termination signal (red colors in Fig. 2). The actual occupation of Pol III on the nc886 genomic region was consistently shown by chromatin immunoprecipitation followed by ultra-high-throughput sequencing (ChIP-seq data) from several research groups [15,16,17]. It is generally thought that Pol III activity is elevated in cancer [18]. In agreement with this idea, the nc886 expression level is high in many cancer cells relative to normal tissues (YSL, unpublished data) [5].
On the contrary, nc886 expression is silenced by CpG DNA hypermethylation in many malignancies [8,9,19,20]. Methylation at CpG dinucleotides is the most common type of epigenetic modification of the mammalian genomic DNA (reviewed in Klose and Bird [21]). Some genes have CpG islands, which are blocks of sequence where CpG dinucleotides are enriched and located mostly in the promoter region. Hypermethylation in CpG islands represses gene transcription and this is one of the most frequent ways that tumor suppressor genes are silenced in cancer. Noticeably, the genomic region of nc886 has a CpG island (blue bar in Fig. 2). Many studies, done mostly on protein-coding genes transcribed by Pol II, indicate that chromatin remodeling is the prevalent mechanism by which CpG methylation silences gene expression. However, CpG methylation has barely been examined in ncRNA genes transcribed by Pol III. To the best of our knowledge, nc886 is the first and so far only case of a Pol III transcript that is silenced by CpG hypermethylation in cancer. So, nc886 should serve as an ideal model gene for studying a mechanism for epigenetic regulation of Pol III transcription.
nc886 is encoded on human chromosome 5q31 and its locus is flanked by transforming growth factor β-induced (TGFBI) and SMAD5, both of which are implicated in the transforming growth factor β (TGF-β) signaling pathway (Fig. 2). Although this suggests a possible co-regulation of nc886 with these two genes, it is interesting to consider the CCCTC-binding factor (CTCF), a barrier protein blocking the spread of heterochromatin (reviewed in Ong and Corces [22]). ChIP-seq data from the Encyclopedia of DNA Elements (ENCODE) indicate that there are two CTCF binding sites at each side of the nc886 region (brown bars in Fig. 2), suggesting that the chromatin status of nc886 is being insulated by CTCF. According to our unpublished data, nc886 seemed to be co-regulated with its flanking genes in some cancer cells but not in others. Given the established role of the TGF-β signaling in cancer, it will be interesting to investigate the involvement of TGF-β in the regulation of nc886/TGFBI/SMAD5 expression.
nc886: A Protein Kinase R Inhibitor ncRNA
As aforementioned, nc886 is a molecular switch for a protein to modulate its activity. That protein is protein kinase R (PKR). PKR is an interferon-inducible, double-stranded RNA (dsRNA)-dependent protein kinase that senses viral infection in the cellular cytoplasm (reviewed in Garcia et al. [23]). The PKR signaling pathway is very complicated: it is activated by several other stresses besides viral infection and its activation affects many downstream pathways. Elaboration on the whole PKR pathway is beyond the scope of this review, but here a very brief introduction only about relevant parts will be provided.
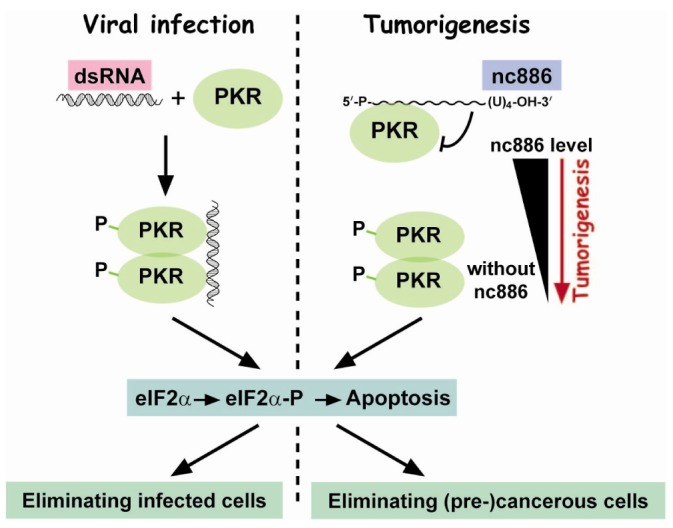
Upon viral infection, the classical PKR pathway begins with its binding to virus-derived dsRNA that activates PKR and subsequently its downstream events, including (1) phosphorylation of eukaryotic initiation factor 2α subunit (eIF2α) to block global protein synthesis and (2) activation of nuclear factor κ-light-chain-enhancer of activated B cells (nuclear factor κB [NF-κB]). The PKR:eIF2α branch leads to apoptosis and this explains how mammalian organisms eliminate virally-infected cells. In contrast, the PKR:NF-κB branch is usually considered to be pro-survival. Besides the innate immune response, numerous papers have also documented PKR's role in cancer in the absence of viral infection. Along with its two opposite functions through eIF2α and NF-κB, both tumor suppressive and oncogenic roles for PKR have been reported in cancer (reviewed in Marchal et al. [24]).
nc886 binds to PKR with an affinity comparable to dsRNA and prevents PKR from being activated, in contrast to the PKR-activating ligand dsRNA [4,5,8,9,10]. One reason for these contradictory outcomes between nc886 and dsRNA is their difference in length. dsRNA is long enough to accommodate PKR dimerization which is a necessary step for its autophosphorylation and subsequent activation (Fig. 3). nc886 at 101 nts long is probably too short for two PKR molecules to bind. When nc886 is ectopically expressed, it attenuates dsRNA-mediated PKR activation [10]. Notably, knockdown of nc886 is sufficient for PKR activation in the absence of dsRNA [4,5,8,9], indicating that human PKR has an intrinsic tendency to self-activate and so needs a repressor to avoid its aberrant activation. In summary, nc886 is a PKR inhibitor.
nc886: Its Tumor Sensing Role
Both knockdown of nc886 and introduction of dsRNA activate PKR (Fig. 3). nc886 silencing does occur during tumorigenesis and dsRNA is generated during viral infection. The significance of PKR in viral infection is to sense viruses and remove them by inducing apoptosis through eIF2α phosphorylation. We implanted this scenario in cancer and proposed a tumor surveillance model. In this model, nc886 silencing in pre-malignant cells is sensed by PKR, which eliminates them through eIF2α phosphorylation. Another important point in this model is the escape of viruses or cancer cells from the PKR:eIF2α cell death pathway. Viruses have various strategies to bypass or disarm PKR. Likewise, some potential cancer cells can survive the PKR surveillance through genetic or epigenetic alterations. For an elaborate description on the tumor surveillance model, please read this review [25].
nc886: Its Tumor Suppressor Role
In addition to the nc886:PKR pathway in the tumor surveillance, does nc886 silencing play an active role in tumor etiology or progression? Two recent papers have provided several pieces of evidence for nc886 being a putative tumor suppressor [8,9].
First, nc886 expression is suppressed in clinical tumor samples compared to normal tissues, and more importantly, the low expression of nc886 correlates with poor survival of patients. This was seen in paired normal/tumor specimens from 88 gastric cancer (GC) and 84 esophageal squamous cell carcinoma (ESCC) patients [8,9]. nc886 expression is also silenced in a number of cancer cell lines of diverse tissue origins, such as esophagus [8], stomach [9], breast (YSL, unpublished data), ovary (YSL, unpublished data), bile duct [4], head and neck, lung, cervix [5], etc. Besides our data, several other research groups have reported that the suppressed expression of nc886 in many different cancer types including lung cancer [19,26,27], thyroid cancer [28], and breast cancer [29]. Although these papers claimed that they detected mature miRNAs, miR-886-5p and -3p, nc886 is most probably what they detected, because their detection methods could not efficiently distinguish the mature miRNAs from nc886.
Second, the silencing of nc886 RNA expression is caused by CpG hypermethylation, which is one of the most common mechanisms to inactivate tumor suppressor genes in cancer. The significance of nc886 CpG methylation in the prognosis of cancer patients has been proven by our ESCC and GC data [8,9] as well as other groups' data in lung cancer [19] and acute myeloid leukemia [20].
Third, acute depletion of nc886 activates oncogenes. These oncogenes include renowned ones, such as FOS, MYC, MAFB, and ID2, all of which are known to drive cells to malignancy when aberrantly overexpressed [30,31,32,33]. Also, nc886 depletion activates oncogenic NF-κB in a PKR-dependent manner [4,8,9] and in this case PKR acts as an oncogene in the few cells that have survived the nc886: PKR:eIF2α-mediated apoptotic branch. In this context, nc886 is an inhibitor of the PKR oncogene and therefore is a tumor suppressor.
Fourth, nc886 is antiproliferative. Ectopic expression of nc886 inhibits growth of several cancer cells [8,9]. This effect is specific, because such toxic effects are seen only in cancer cells that do not express endogenous nc886 but not in non-malignant cells that express endogenous nc886. nc886's antiproliferative role is thought to be, at least partially, through suppressing NF-κB, as supported by our experimental data from cholangiocarcinoma [4].
Conclusion and Future Direction
nc886 is a newly discovered ncRNA that controls PKR activity and plays tumor sensing and suppressing roles. nc886's expression pattern and function in tumors opens a possibility that its measurement can be a diagnostic or prognostic marker and its administration into cancer cells can be a therapeutic strategy. Nonetheless, we expect that nc886's role is multifaceted in cancer and may even be oncogenic in certain contexts, because nc886 expression as well as PKR's role can be in two opposite directions in cancer. Also, nc886 is a unique case of a Pol III gene that is epigenetically silenced in cancer and so provides an intriguing model for studying epigenetic regulation of Pol III.