Membrane Proteins Involved in Epithelial-Mesenchymal Transition and Tumor Invasion: Studies on TMPRSS4 and TM4SF5
Article information
Abstract
The epithelial-mesenchymal transition (EMT) is one mechanism by which cells with mesenchymal features can be generated and is a fundamental event in morphogenesis. Recently, invasion and metastasis of cancer cells from the primary tumor are now thought to be initiated by the developmental process termed the EMT, whereby epithelial cells lose cell polarity and cell-cell interactions, and gain mesenchymal phenotypes with increased migratory and invasive properties. The EMT is believed to be an important step in metastasis and is implicated in cancer progression, although the influence of the EMT in clinical specimens has been debated. This review presents the recent results of two cell surface proteins, the functions and underlying mechanisms of which have recently begun to be demonstrated, as novel regulators of the molecular networks that induce the EMT and cancer progression.
Epithelial-Mesenchymal Transition
Metastasis is the leading cause of cancer-related deaths in most cancer types. As an initial step in cancer metastasis, epithelial tumor cells in general disseminate from a primary solid tumor mass and invade into the surrounding stromal tissues. Invasion is enhanced by tumor cell activation of the epithelial-mesenchymal transition (EMT) [1, 2, 3, 4]. The EMT is characterized by the loss of epithelial apicobasal polarity and cell-cell contacts, modulation of cell-matrix adhesion, enhanced proteolytic activity, cytoskeletal remodeling, and acquisition of the ability to migrate and invade the extracellular matrix (ECM) [1, 3]. During the EMT, epithelial cells undergo molecular changes; epithelial cells gradually lose their epithelial markers, such as E-cadherin, ZO-1, and cytokeratins, and concomitantly acquire mesenchymal markers, such as vimentin, fibronectin, N-cadherin, and alpha smooth muscle actin [1, 3]. The EMT plays a critical role in the formation of various tissues and organs, such as the mesoderm, neural crest, heart, secondary palate, and peripheral nervous systems, during embryonic development and wound healing in adult organism [2, 4]. Furthermore, the EMT is implicated in pathological processes, such as tumor cell invasion and metastasis and organ fibrosis [2].
One of the hallmarks of the EMT is the functional loss of E-cadherin, which is currently thought to be a metastasis suppressor [5]. Downregulation of E-cadherin is usually mediated by E-cadherin transcriptional repressors/EMT-inducing transcription factors, including the Snail superfamily of zinc-finger factors (Snail and Slug), the ZEB family (ZEB1 and ZEB2), and basic helix-loop-helix factors (Twist1 and E47), which have been associated with tumor invasion and metastasis [4, 5]. These factors repress transcription of E-cadherin by interacting with proximal E-box elements in the E-cadherin promoter [5]. In addition, these E-cadherin repressors may be directly or indirectly involved in the upregulation of certain mesenchymal genes [5], although the precise mechanism of this regulation is largely unknown.
The EMT is triggered by soluble growth factors, such as members of the transforming growth factor-β (TGFβ) and fibroblast growth factor families, epidermal growth factor, and hepatocyte growth factor (HGF) [3, 4]. Subsequent activation of receptor-mediated signaling triggers the activation of intracellular effector molecules, such as members of the small GTPase family, leading to changes in cytoskeletal organization, and also results in the activation of EMT-inducing transcription factors [3, 4]. In addition, components of the ECM, such as collagen, and activation of integrin co-receptors are also involved in the EMT process [3]. Certain proteases are sufficient to induce the EMT [2]; for example, matrix metalloproteinase-3 triggers the EMT by increasing the cellular levels of reactive oxygen species, which in turn induces Snail expression [6].
Recently, microRNAs (miRs) have been identified as a novel class of EMT regulators; miRs that negatively regulate the EMT include miR-153, -155, -194, -25, -212, and -200, and miRs that positively regulate the EMT include miR-29a, -103/107, -150, and -221/22 [7]. miRs regulate invasion and metastasis by targeting the transcripts of various genes involved in the EMT event, including EMT-inducing transcription factors. For example, members of the miR-200 family are negative regulators of the EMT and are essential for maintenance of the epithelial status through the downregulation of ZEB1 and ZEB2. In turn, miR-200 members are transcriptionally repressed by ZEB1 and ZEB2, thus establishing a double-negative feedback loop [8].
The EMT was recently shown to be linked to stemness and self-renewal capacity [9, 10]. In cases of breast cancer stems, the linkage among EMT phenotype, stemness, and drug resistance has been well studied [11]. Further, epithelialmesenchymal plasticity (consisting of EMT and MET) is also described in circulating tumor cells (CTCs) [12, 13, 14]. CTCs with various degrees of EMT phenotypes are found during breast cancer metastasis [15]. Therefore, CTCs may involve self-renewal capacity, which is linked to the EMT, during cancer metastasis [16].
Transmembrane Protease Serine 4 (TMPRSS4)
Introduction to type II transmembrane serine proteases
Dysregulation of proteases is a hallmark of cancer progression; thus, proteases in general have been the subject of numerous cancer studies. Extracellular proteolytic enzymes, including matrix metalloproteinases (MMPs) and serine proteases, contribute to tumor cell invasion and metastasis through both direct proteolytic activity and the regulation of cellular signaling and functions [17, 18, 19]. Most members of the serine protease family are either secreted or sequestered in cytoplasmic organelles awaiting signal-regulated release. Recently, type II transmembrane serine proteases (TTSPs) have been recognized as a new subfamily of serine proteases that have in common an extracellular proteolytic domain, a single-pass transmembrane domain, a short intracellular domain, and a variable-length stem region containing modular structural domains [20, 21, 22, 23, 24]. Enteropeptidase (also known as enterokinase), identified over a century ago due to its pivotal role in food digestion, is the first TTSP, which was revealed by molecular cloning of the enteropeptidase cDNA two decades ago [25]. TMPRSS2, human airway trypsin-like protease (HAT), corin, and matriptase have been subsequently identified as cell surface-associated proteases [23, 24]. To date, 20 TTSPs have been identified in mouse and humans due to the analysis of sequence data from mammalian genome projects [23]. Analysis of the tissue distribution of TTSPs and gene targeting in mice of certain TTSPs suggested that a significant number of TTSPs may have important functions in embryonic development and homeostasis of mammalian tissues, such as heart, skin, inner ear, placenta, and digestive tract [23, 24].
Most TTSPs are overexpressed in a variety of tumors compared to normal tissues, implicating their potential as novel markers of tumor development and progression and possible molecular targets for anti-cancer therapeutics [23, 26]. Recently, a number of works have focused on the evaluation of the expression of individual TTSPs during tumor progression and on the investigation of the potential roles of these proteases in tumor cell proliferation, migration, and invasion [23, 27].
TMPRSS4 in cancer
TMPRSS4 (gene ID, 56649; chromosomal location, 11q23.3), initially referred to as TMPRSS3, was originally identified as a gene expressed in most pancreatic cancer tissues but not in the normal pancreas or chronic pancreatitis [28]. To date, 7 isoforms have been reported. The deduced sequence of 437 amino acids of the longest isoform (isoform 1) contains a serine protease domain with putative trypsin-like activity and a transmembrane domain [28]. In human, TMPRSS4 mRNA was detected in bladder, esophagus, stomach, small intestine, colon, and kidney [28], although the physiological roles of TMPRSS4 remain unknown. Furthermore, TMPRSS4 expression was upregulated in malignant compared to benign thyroid neoplasm and was suggested as both a diagnostic and prognostic marker [29, 30]. TMPRSS4 was associated with poor prognosis in non-small-cell lung cancer (NSCLC) with squamous cell histology [31], triple-negative breast cancer [32], cervical cancer [33], and gastric cancer patients [34]. Kim et al. [35] reported that TMPRSS4 mRNA levels were upregulated in colorectal cancer tissues versus adjacent normal mucosa. The authors also reported that TMPRSS4 protein expression was significantly higher in human colorectal cancer tissues from advanced stages (52.5% and 50.0% of stages III and IV, respectively) than in that of early stage (6.3% in stage I), suggesting that TMPRSS4 may play a role in the progression of non-invasive tumors to invasive malignancies [35]. Jia et al. [36] showed that the inhibitory tripeptide tyroserleutide led to downregulation of TMPRSS4 in hepatocellular carcinoma (HCC), thereby reducing the invasion and metastasis of HCC induced by irradiation. Taken together, TMPRSS4 may be a novel biomarker for the prognosis of certain types of cancers and could be employed for diagnostics and therapeutics.
On the other hand, the mechanism by which TMPRSS4 expression is modulated has not been well characterized. Recently, Nguyen et al. [37] reported that TMPRSS4 was increased in NSCLC cells under hypoxic conditions, suggesting that hypoxia within the tumor microenvironment may upregulate TMPRSS4 expression.
Function of TMPRSS4 in the regulation of EMT and invasion
In colon cancer cells, TMPRSS4 induces downregulation of E-cadherin and leads to EMT events, accompanying morphological changes and actin reorganization [38]. Suppression of TMPRSS4 by siRNA reduces cell invasion in colon and lung cancer cells, while overexpression TMPRSS4 induces migration, invasion, and metastasis [38]. Attachment and spreading of cells on the ECM, with concomitant formation of stress fibers and focal adhesions, is a prerequisite for cell migration. TMPRSS4 also modulates cell-matrix adhesion and cell spreading mainly through modulation of integrins, such as α5β1, which has been centrally implicated in the EMT and cell motility [39, 40], which probably contributes to enhanced motility and invasiveness. One of the molecular mechanisms by which TMPRSS4 mediates the EMT and invasiveness in tumor cells is that TMPRSS4 mediates focal adhesion kinase (FAK) signaling pathway activation and extracellular signal-regulated kinase (ERK) activation, mainly through integrin α5 upregulation, leading to the EMT and invasiveness. Furthermore, TMPRSS4 overexpression in human colorectal cancer tissues positively correlates with enhanced expression of integrin α5 and inversely correlates with E-cadherin expression, confirming that TMPRSS4 modulates expression of EMT markers. Recently, Larzabal et al. [41] reported that miR-205 is involved in TMPRSS4-induced integrin α5 expression in NSCLC cells. To further implicate TMPRSS4 in the EMT, Cheng et al. [42] suggested that interactions between HGF activator inhibitor (HAI-1) and TMPRSS4 contribute to EMT events, including E-cadherin reduction and morphological changes in pancreatic cancer cells. In addition, TMPRSS4-induced E-cadherin reduction and EMT play a critical role in radiation-induced long-term metastasis of residual HCC in nude mice [43].
Interaction of TMPRSS4 and integrin α5, based on the observation that TMPRSS4 partially interacts with integrin α5 under certain coimmunoprecipitation conditions in a cell line-dependent manner [35] (Kim S, unpublished observation), suggests the possibility that TMPRSS4 may modulate or participate in the interaction of integrin and other cell surface proteins (for example, tetraspanin, receptor tyrosine kinases, etc.), leading to subsequent signaling transduction activation. In fact, TMPRSS4 can interact with urokinase plasminogen activator receptor (uPAR; CD87) [44], which can induce the EMT in hypoxic breast cancer cells [45], although it is not clear whether TMPRSS4 interacts with uPAR directly or via integrin(s).
Loss or reduction of E-cadherin expression is a well-known hallmark of the EMT and correlates positively with tumor cell invasion and metastasis [3]. TMPRSS4 appears to modulate SIP1/ZEB2 expression, based on the observation that SIP1 mRNA is upregulated in TMPRSS4-overexpressing colon cancer cells, although induction of SIP1 at the protein level remains to be determined. Therefore, it is possible that SIP1 mediates TMPRSS4-induced EMT events, including E-cadherin reduction.
Several studies have shown that suppression of high endogenous E-cadherin expression renders non-invasive cells partially invasive [46], whereas reconstitution of E-cadherin results in tumor cell reversion from an invasive mesenchymal phenotype to a benign epithelial phenotype [46, 47]. In contrast, other studies have shown that ectopic expression of E-cadherin could not reverse EMT phenotypes induced by the transcription factor Twist1 [10]. On the other hand, downregulation of E-cadherin was required for TMPRSS4-mediated EMT and invasion in colon cancer cells but was not sufficient for induction of these phenotypes [35], suggesting that downregulation of E-cadherin is not the sole contributor to TMPRSS4-mediated phenotypes. In this respect, upregulation of specific mesenchymal markers, such as integrin α5, besides the downregulation of E-cadherin by TMPRSS4, may be required for full invasiveness during colon cancer progression (Fig. 1).
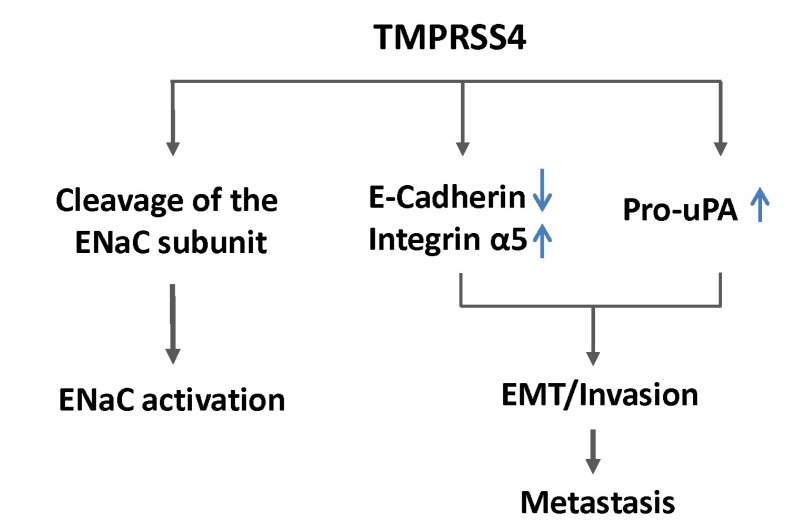
Cellular functions of TMPRSS4. ENaC, epithelial sodium channel; uPA, urokinase plasminogen activator; EMT, epithelialmesenchymal transition.
Molecular mechanisms and signals regulated by TMPRSS4
Numerous studies have focused on the investigation of in vivo substrates of TTSPs. However, few studies have conclusively addressed the in vivo molecular targets and function of TTSPs during tumor progression. In vitro, several TTSPs, including matriptase, were shown to activate prourokinase plasminogen activator, pro-macrophage stimulating protein-1, and pro-HGF, which are implicated in the proliferation, migration and invasion of various cancer cell types [23].
Like most of the members of the TTSP family, TMPRSS4 can activate epithelial sodium channel (ENaC) in vitro through its proteolytic activity, possibly regulating sodium and water flux across high-resistance epithelia [48, 49]. TMPRSS4 induces cancer cell invasion in a manner that is dependent on serine proteolytic activity [38], and inhibitory compounds against TMPRSS4 serine protease activity were reported to reduce colon cancer cell invasion [50]. However, it remains unknown which precursor substrates are cleaved by TMPRSS4 to contribute to tumor progression. On the other hand, it has recently been reported that TMPRSS4 induces urokinase plasminogen activator (uPA) gene expression through activation of the transcription factors AP-1, Sp1, and Sp3 in mainly a JNK-dependent manner in prostate and lung cancer cells but not in colon cancer cells [44]. uPA is a well-known serine protease involved in invasion and metastasis and correlates with poor prognosis in breast, lung, stomach, bladder, colon, prostate, and ovarian cancers [51], and TMPRSS4 expression significantly correlates with uPA expression in human lung and prostate adenocarcinomas [44]. In addition, TMPRSS4-mediated uPA expression contributes to prostate cancer cell invasion (Fig. 1) [44]. It is intriguing that TMPRSS4 activates JNK signaling pathways, possibly through its association with uPAR, leading to uPA expression. uPAR can induce the EMT and stem cell-like properties in breast cancer cells by activating diverse cell signaling pathways, including ERK, phosphoinositide-3-kinase-Akt, and Rac1 [45, 52]. Therefore, the association of TMPRSS4 and uPAR and subsequent cell signaling modulation may be a novel mechanism for the control of invasion and the EMT.
The observations that TMPRSS4 modulates cell signaling and subsequently activates both AP-1 and Sp1/3 transcriptional activities [44], which have been reported to be involved in the transcriptional regulation of the EMT and invasion [53], suggest that TMPRSS4 could modulate the expression of various genes, which may be associated with invasion and metastasis.
Transmembrane 4 L Six Family Member 5 (TM4SF5)
The tetraspanins
Tetraspanins (TM4SFs) have four transmembrane protein domains with two extracellular loops and one intracellular loop (ICL) and the N- and C-terminal tails [54]. They are expressed on the cell surface and/or intracellular vesicles and contain 33 members in mammals [55]. Tetraspanins, or TM4SFs, are suggested to be located at tetraspanin-enriched microdomains (TERMs) [56], where they form protein-protein complexes in a homophilic or heterophilic manner with other TM4SFs, integrins, or growth factor receptors [57, 58]. The protein complexes are known to regulate the dynamics of the complex components on the cell surface with regard to diffusion, trafficking, retention, and stability, in addition to influencing intracellular signal transductions [56, 59, 60].
TM4SF5 in cancer
TM4SF5 (gene ID, 9032) maps to chromosome 17 at 17p13.3 according to Entrez Gene. In AceView, it covers 11.34 kb, from 4621928 to 4633262 (NCBI 36, March 2006), on the direct strand containing 4 different gt-ag introns. Its transcription produces 2 alternatively spliced mRNAs via alternative polyadenylation sites, which putatively encode 2 different isoforms (197 and 132 amino acids), containing an L6 membrane domain (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=35g&c=Gene&l=TM4SF5). TM4SF5 (20,823 Da) is a transmembrane glycoprotein; as a family group, it is related to the tetraspanin family (transmembrane 4 L six family), including TM4SF1 (L6, L6-Ag), TM4SF4 (IL-TIMP), TM4SF518 (L6D), and TM4SF20 [61, 62]. TM4SF5 is highly expressed in diverse types of cancers, including liver, pancreatic, gastric, colon, adrenocorticotropic hormone (corticotropin)-negative bronchial carcinoid tumors, soft-tissue sarcoma, nonendocrine lung, and papilla vateri carcinoma [63, 64, 65, 66]. Similar to tetraspanins (i.e., transmembrane 4 superfamily, TM4SFs), TM4SF5 has four transmembrane domains (TM1-TM4), short cytoplasmic domains at their N- and C-termini, an ICL between TM2 and TM4, two extracellular loops, a smaller extracellular loop between TM1 and TM2, and a larger extracellular loop between TM3 and TM4 [61, 62]. Recent clinical studies separately report that TM4SF5 is highly expressed in tumors from deceased breast cancer patients, compared to those from 10-year breast cancer survivors [67], and that postoperative 5-year overall survival of esophageal cancer patients negatively correlates with TM4SF5 expression [68]. These reports suggest that TM4SF5 overexpression correlates with poor prognosis of cancer patients.
TM4SF5-mediated regulation of signaling molecules
TM4SF5 can appear to form TERMs on the cell surface, via formation of large protein-protein complexes with tetraspanins, integrins, and growth factor receptors [61, 69]. Therefore, by virtue of the protein complex formation, overexpressed TM4SF5 in cancer cells can influence or activate diverse intracellular signaling pathways for cell adhesion, proliferation, the EMT, migration, and invasion for tumor progression and maintenance.
TM4SF5 is shown to associate with integrins α2, β1 [70, 71], α5 [72], and epidermal growth factor receptor (EGFR) [73, 74] during cell migration [70, 71], angiogenesis [72], drug resistance [74], and fibrosis [73]. With association and retention of integrin α5 on the cell surface, TM4SF5 can activate intracellular signaling for FAK/c-Src activation, leading to STAT3 activity for vascular endothelial growth factor (VEGF) induction [72]. In addition, TM4SF5 directly interacts with FAK or c-Src to regulate migration [75] and invasive ECM degradation [76]. In addition, TM4SF5 expression causes AKT activation, which in turn causes phosphorylation of p27Kip1 Ser10 for its cytosolic translocation, where it can regulate RhoA activity for morphological change and migratory function [74].
TM4SF5-mediated EMT in tumor progression
TM4SF5 expression in hepatocytes or NSCLC leads to EMT phenotypes, which in turn cause loss of contact inhibition [74], enhance migration and invasion for metastasis [77], and render gefitinib resistance [78]. TM4SF5 expression causes morphological changes through abnormal regulation of RhoA and Rac1 in hepatocytes, together with the loss of E-cadherin expression leading to EMT induction [74] via induction of Slug [79]. Inhibition of TM4SF5-mediated signaling events of the cytosolic enrichment of p27Kip1 abolishes abnormal multilayer cell growth [74] and retards the G1 to S phase progression [80]. Further, inhibition of TM4SF5-mediated EMT by suppression of cytosolic p27Kip1 expression leads gefitinib-resistant NSCLC cells to become gefitinib-sensitive [78]. TM4SF5 is involved in activation of hepatic stellate cells by causing EMT processes, leading to a correlation with the development of liver fibrosis in CCl4-treated mouse models [81]. TM4SF5 expression is achieved by TGFβ1-mediated Smad actions on EGFR activation [73], such that the important roles of the multifunctional cytokine TGFβ1 in the activation of hepatic stellate cells and the EMT are confirmed in a development of murine liver fibrosis. Since liver fibrosis can eventually lead to hepatocarcinoma at a high rate of over 70% [82], the roles of TM4SF5 in the development of both fibrosis and tumorigenesis in the liver can be reasonable.
Meanwhile, TM4SF5 expression enhances directional migration and invasion of hepatocytes. TM4SF5 in hepatocytes causes directional migration at an enhanced speed and the formation of more invadosome-like structures enriched with cortactin, actin, and actin-regulatory proteins, like Arp2 and WASP [77]. TM4SF5-mediated directional migration involves direct interaction and activation of FAK via the ICL domain of TM4SF5 and the F1 lobe of the FAK FERM domain [75]. Further, TM4SF5-mediated invasive ECM degradation requires direct interaction between the COOH-terminus of TM4SF5 and c-Src, which is linked to Tyr845 phosphorylation of EGFR to form more invasive protrusions [76]. TM4SF5-mediated multilayer growth [74], FAK activity, migration, and invasion [75] are abolished by an anti-TM4SF5 reagent, TSAHC (a synthetic compound), which appears to affect its N-glycosylation and at the same time block induction of the TM4SF5-dependent EMT phenotype and multilayer growth [83]. Therefore, TM4SF5 also plays important roles in tumor initiation and progression, possibly being supported by an EMT process.
Other TM4SF5-mediated EMT-related biological processes
The EMT is well known to be related also to the development [84] and stemness of self-renewal [9]. We also observed that TM4SF5 can play roles in other EMT-mediated biological processes, like development of muscles and self-renewal of cancer cells. In zebrafish, suppression of tm4sf5 results in abnormal development, with an aberrant trunk and morphology of muscle fibers, presumably via alterations in the expression and localization of integrin α5, which is necessary for somite boundary maintenance (Choi YJ and Lee JW, in revision). In addition to liver fibrosis and tumorigenesis, therefore, TM4SF5 expression is importantly involved in the development of zebrafish muscles, which might bemediated by EMT.
Presumably, these diverse cellular effects by TM4SF5 expression might be possible due to the characteristics of TM4SF5-similar to tetraspanins-which forms large protein networks via heterophilic or homophilic interactions between tetraspanins, integrins, and growth factor receptors. TM4SF5 is shown to bind integrin α2, β1 [70, 71], α5 [72], EGFR [73], and interleukin 6 receptor (IL6R) (Ryu J and Lee JW, in revision). Although its ligand has not been identified, interaction(s) with (an)other membrane protein or receptor can recapitulate the ligand binding-based activation. Therefore, TM4SF5 can transduce signaling activities for diverse cellular functions, including the EMT and different EMT-mediated phenotypes. Although diverse miRs are known to regulate the EMT [7], miRs targeting TM4SF5 are being studied.
TM4SF5-mediated gene regulation
Comparison of protein expression patterns between TM4SF5-null and -expressing cells shows a negative correlation between TM4SF5 and cell-cell adhesion-related molecules of epithelial markers, including E-cadherin [74], and a positive correlation between TM4SF5 and mesenchymal markers, including Slug [79], supporting TM4SF5-mediated EMT. Reverse transcription polymerase chain reaction analyses of show that TM4SF5-mediated regulation of their expressions occurs at the transcriptional level (Lee JW, unpublished observation). However, the signaling pathways underlying this regulation have not been determined yet.
In addition, TM4SF5 expression also correlates with cytosolic p27Kip1 [74]. Although p27Kip1 in the nucleus is inhibitory to cyclin-dependent kinases, suppressing the cell cycle and proliferation, its localization in the cytosol can lead to tumorigenic functions [85]. Cytosolic p27Kip1 has been reported in different clinical reports, where different cancer types show enriched cytosolic localization of p27Kip1 [86, 87, 88], suggesting that cytosolic p27Kip1 can be tumorigenic [89]. p27Kip1 can be phosphorylated by Akt, KIS, or JNK [90, 91, 92], resulting in translocalization and stabilization in the cytosol, where it binds to and inactivates RhoA GTPase, leading to alterations in actin organization and motility regulation [93]. TM4SF5 expression also causes overexpression of p27Kip1, although how it occurs is unknown; TM4SF5 causes Akt-mediated Ser10 phosphorylation of p27Kip1, leading to its stabilization, RhoA activity changes, and eventually morphological elongation for the EMT and contact inhibition loss [74]. JNK-mediated p27Kip1 phosphorylation in a TM4SF5-dependent manner also results in localization of p27Kip1 at cell-cell contacts [91], possibly leading to altered actin organization at cell-cell contacts. In addition, inhibition of the proteasome in terms of activity and subunit expression also depends on TM4SF5 expression, resulting in morphological changes and the EMT, suggesting another novel mechanism for TM4SF5-mediated EMT [79].
Meanwhile, TM4SF5 causes activation of the FAK/c-Src signaling pathways, leading to STAT3 phosphorylation at Tyr705 for the induction and secretion of VEGF, which can stimulate neighboring endothelial cells for enhanced (tumor) angiogenesis [72]. During modeling of the tumor microenvironment, cancer cells overexpressing TM4SF5 appear to negatively regulate expression of the cytokine IL6, and exogenous IL6 treatment leads to less STAT3 signaling activation in TM4SF5-positive cancer cells (Ryu J and Lee JW, in revision); thus, TM4SF5-dependent suppression of IL6 can be a strategy for TM4SF5-positive tumor cells to avoid pro-immunological actions by IL6 secreted by neighboring immune cells. As for invasion, TM4SF5 expression also increases the mRNA and protein levels of MMP2, in addition to its activity [77].
Therefore, TM4SF5 expression correlates with or plays important roles in tumorigenesis in different mechanisms, including induction of the EMT and gene regulation (Fig. 2).

TM4SF5-mediated epithelial-mesenchymal transition (EMT) is involved in diverse cellular functions, leading to liver tumorigenesis and maintenance in addition to developmental processes.
Concluding Remarks
Considering that such membrane proteins as TMPRSS4 or TM4SF5 may be important upstream regulators of the EMT and the invasiveness of cancer cells and because their expression differs substantially in normal and cancer tissues, targeting them could be a novel therapeutic strategy for the treatment of cancer metastasis. In the future, the functional involvement of TMPRSS4 and/or TM4SF5 in the initiation and progression of tumors needs to be evaluated using mouse models. Cancer-associated mutations and singlenucleotide polymorphisms within TMPRSS4 or TM4SF5 also need to be analyzed in association with cancer risk.