A Follow-up Association Study of Genetic Variants for Bone Mineral Density in a Korean Population
Article information
Abstract
Bone mineral density (BMD) is one of the quantitative traits that are genetically inherited and affected by various factors. Over the past years, genome-wide association studies (GWASs) have searched for many genetic loci that influence BMD. A recent meta-analysis of 17 GWASs for BMD of the femoral neck and lumbar spine is the largest GWAS for BMD to date and offers 64 single-nucleotide polymorphisms (SNPs) in 56 associated loci. We investigated these BMD loci in a Korean population called Korea Association REsource (KARE) to identify their validity in an independent study. The KARE population contains genotypes from 8,842 individuals, and their BMD levels were measured at the distal radius (BMD-RT) and midshaft tibia (BMD-TT). Thirteen genomic loci among 56 loci were significantly associated with BMD variations, and 3 loci were involved in known biological pathways related to BMD. In order to find putative functional variants, nearby SNPs in relation to linkage equilibrium were annotated, and their possible functional effects were predicted. These findings reveal that tens of variants, not a single factor, may contribute to the genetic architecture of BMD; have an important role regardless of ethnic group; and may highlight the importance of a replication study in GWASs to validate genuine loci for BMD variation.
Introduction
In an aging society, osteoporosis is a major health problem. It is characterized by systemic impairment of bone mass, strength, and microarchitecture, leading to increased risk of fragility fractures, particularly hip fracture [1]. In the United States, the cost of incident fractures due to osteoporosis and low bone mass was $17 billion in 2006. It is expected to rise to around $25.3 billion by the year 2025 [2]. Likewise, the burden for them is prevalent in Asia. The annual incidence of osteoporosis-related fractures increased to 1,661, 1,646, 1,623, and 1,614 per 100,000 persons from 2005 to 2008 in Korea [3]. Osteoporosis is defined by the measurement of bone mineral density (BMD), which is so far the best single predictor [4, 5].
BMD is a genetic component, with estimates of heritability ranging from 0.46 to 0.84 at all skeletal sites [6]. In recent years, genome-wide association studies (GWASs) have enhanced the understanding of the genetic architecture underlying quantitative traits and common, complex diseases [7]. These studies provide key insights into the variation of quantitative traits and prospects for designing specifically effective strategies for diseases [8]. There have been many GWASs to identify genetic loci that influence BMD, and dozens of candidates have been presented [9,10,11,12,13]. However, only several loci have been consistently identified in a few replication studies [14,15,16], suggesting that BMD is a complex trait affected by multiple components [17].
Recently, a meta-analysis of 17 studies for BMD of the femoral neck and lumbar spine was performed for the first time in order to identify consensus BMD-associated single-nucleotide polymorphisms (SNPs) throughout races [18]. It included 32,951 individuals across North America, Europe, East Asia, and Australia. The meta-analysis offered 56 associated loci that were highly significant in both the discovery stage and replication stage. This also proposed a model of BMD loci and successfully validated it with an independent population.
Here, we investigate those 56 BMD loci in a Korean population called Korea Association REsource (KARE) [19]. The analysis showed that BMD variation is a polygenic and complex trait, and it could be even different, depending on sex and the skeletal site. The finding that some crucial loci are still significant in KARE supports the validity of BMD loci. This study may contribute to the importance of a replication study in GWASs to understand the common mechanism underlying BMD.
Methods
Study subjects
Subjects from the KARE GWAS have been described [19]. Since it launched in 2001, the total of 10,038 participants have been recruited from the Anseong and Ansan cohorts in the Republic of Korea. Both cohorts were designed to view a variety of quantitative traits from a long-term perspective. Besides, clinical surveys have been performed through epidemiological surveys and physical examinations. Participants have been examined every 2 years, with a third follow-up study in 2008. The analysis of KARE data has been approved by the Korea National Institute for Bioethics Policy (KONIBP), Republic of Korea (P01-201311-RS-02-01).
Genotyping, quality control, and imputation
A total of 10,004 DNA samples were genotyped with the Affymetrix Genome-Wide Human SNP array 5.0 (Affymetrix, Santa Clara, CA, USA). Genotypes were called by Bayesian Robust Linear Modeling using the Mahalanobis Distance (BRLMM) algorithm [20]. The quality score criteria and procedures have been described in a previous GWAS [19]. After each step of the quality control, genotypes from 8,842 individuals with a total of 352,228 markers were left and used for the association analysis. In this study, we additionally excluded 257 individuals taking medication that influenced BMD levels.
SNP imputation was conducted using the IMPUTE2 program [21]. The 1000 Genomes Phase I integrated haplotypes (released in Dec 2013), produced using SHAPEIT2 in NCBI build 37 coordinates, were used as the reference panel [22]. All 1,092 individuals with 36.8 million SNPs were initially utilized, and haplotypes with singleton sites were removed. Since there is no X chromosome in the current version of the reference panel, a previous version (released in Mar 2012) was used as the reference panel for the X chromosome.
Because the previous GWAS was based on NCBI build 36, SNP positions should be converted to build 37 coordinates by using the University of California, Santa Cruz (UCSC) liftOver tool [23]. SNP orientations were aligned to the forward strand using PLINK [24]. In order to improve the performance of the SNP imputation, SNPs were pre-phased by SHAPEIT2 [22, 25]. Then, SNPs were imputed directly onto the haplotype estimates by IMPUTE2. Imputation was carried out for the regions within 500 kb on either side of 64 SNPs reported in the previous GWAS [18]. SNPs with a minor allele frequency < 0.01 and imputation quality score (INFO) < 0.4 were filtered out. Then, we combined 51 imputed SNPs with 9 directly typed KARE SNPs for association analyses.
Statistical analysis
The association analyses were done using PLINK version 1.07 [24] for both genotyped and imputed SNPs. A test for BMD was done by linear regression with an additive model (1 degree of freedom [d.f.]) after adjustments for age, sex, and recruitment area. Statistical significance was determined by p < 0.05 in a two-tailed test. p-values were not adjusted for multiple testing of SNPs. Proximal SNPs and pairwise linkage disequilibrium (LD) were computed by PLINK. A threshold for correlation was set as r2 > 0.8. Annotation of SNPs was done by SNPnexus [26]. The effect of SNPs on protein function was predicted using SIFT [27] and PolyPhen [28]. Transcription factor binding sites were inferred from the TRANSFAC database [29].
Results
The KARE study population included GWA genotypes from 8,842 individuals [19]. We removed 257 individuals currently taking medication to remove any potential influence on BMD. BMD was measured by speed of sound (SOS) at the distal radius (BMD-RT) and midshaft tibia (BMD-TT), but the value was transformed to T-score for analysis. The mean T-score of the radius and tibia of the 8,586 subjects was 0.038 ± 0.016 (SEM) and -0.472 ± 0.017, respectively.
The meta-analysis of 17 GWASs identified 64 SNPs in 56 loci associated with BMD with genomewide significance. The analysis also proposed a model of BMD-decreasing alleles, and their effects were validated with an independent population: a prospective study in postmenopausal Danish women aged 55-86 years. The alleles were scored with weights based on their individual effects on BMD. Even though the previous GWAS was based on lumbar spine and femoral neck BMD, we assumed that the effects of BMD loci would be available to BMD at other sites, such as the distal radius and midshaft tibia. Thus, the model was applied to the KARE population to prove its validity.
Among 64 SNPs, 9 SNPs were genotyped in the chip, but 55 other SNPs were imputed from genotypes in the 1000 Genomes (1KG) Project (Phase I). After filtering, 60 SNPs were used for the analysis, and individuals were scored according to the scoring system of the previous study. Then, individuals were grouped into five bins based on their scores, and the mean of BMD of each group was measured (Fig. 1). While BMD-RT did not follow the model proposed in the previous study, BMD-TT showed the pattern as expected. In other words, BMD at the midshaft tibia decreased as the score increased. The analysis on the basis of 60 SNPs explained 3.2% of the total genetic variance in BMD-TT.

Combined effect of bone mineral density (BMD)-decreasing alleles in Korea Association REsource (KARE) data. The genetic score of each individual was computed on the basis of the weighted model proposed in a previous study [18]. Effects are shown for BMD levels measured at the distal radius (BMD-RT) (A) and BMD levels measured at the midshaft tibia (BMD-TT) (B). Histograms represent the numbers of individuals with each genetic score area (left y-axis). Round spots (right y-axis) show mean BMD-RT standardized values in A and mean BMD-TT standardized values in panel (B). Vertical lines represent 95% confidence limits.
In order to identify variants significantly influential in the Korean population, an association analysis of 60 SNPs was done with KARE data. Single-marker tests of association with BMD were carried out by trend test (1 d.f.) after adjustments for age, sex, and recruitment area (Table 1). Twelve SNPs showed a directionally consistent association with the previous GWAS (p < 0.05). Six loci were associated with BMD-RT and six loci were associated with BMD-TT, but there were no loci associated with both. One of these loci is on the X chromosome. The same analysis was carried out by gender strarification (Table 2). Three loci were prioritized in the female sample set, and one of them (rs13204965) was newly found in the analysis. Therefore, a total of 13 independent SNPs were selected for further analysis (Fig. 2).

Estimated effects of genomewide-significant SNPs on BMD-RT and BMD-TT

Gender-stratified association analysis of SNPs on BMD-RT and BMD-TT

Effects of significant BMD loci on BMD at different skeletal sites. Genetic effects are expressed as standardized values and shown for BMD levels measured at the distal radius (BMD-RT; filled circles) and BMD levels measured at the midshaft tibia (BMD-TT; open circles). Horizontal lines represent 95% confidence limits. ars13204965 was seen as significant in women only.
As mentioned in the previous GWAS, some of these 13 SNPs were seen as members of biological pathways related to BMD: the RANK-RANKL-OPG pathway (TNFRSF11B), mesenchymal stem cell differentiation (SP7), and Wnt signaling (WNT4). Interestingly, SNPs in TNFRSF11B and SP7 were associated with BMD-RT, whereas the SNP in WNT4 was associated with BMD-TT (Tables 1 and 2).
For the purpose of identifying putative functional SNPs, markers in the surrounding 100-Kb region were examined using LD (Table 3). Likewise, they were imputed from 1KG Phase I, centered on 13 loci. Eight BMD-associated SNPs were highly correlated (r2 > 0.8) with functional SNPs annotated in the 1KG reference. rs13245690 was related to 5 putative functional variants, which correlated with each other by LD (r2 > 0.8). Two SNPs (rs1524498 and rs41281692) were located within coding regions in CPED1. rs1524498 leads to synonymous coding, but rs41281692 leads to a conservative change of an amino acid. The remaining 3 SNPs (rs10228519, rs10274486, and rs1357756) were located in the intronic regions in CPED1 and are predicted to alter alleles in transcription factor binding sites. Each allele was anticipated to affect the binding of CUTL1, POU2F1, and GATA-1, respectively.
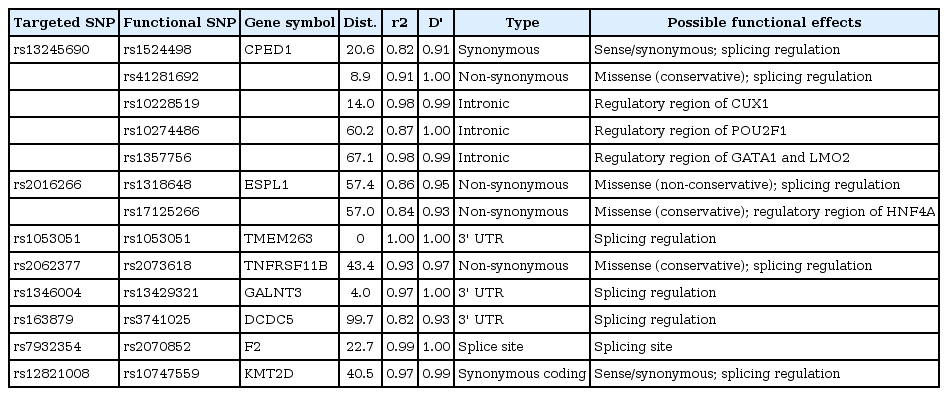
Predicted functional SNPs in LD with targeted SNP in each locus
rs2016266 in SP7 was correlated with two SNPs (rs1318648 and rs17125266) in ESPL1. Both were associated with missense mutations. rs17125266 was predicted to be conservative, but rs1318648 was predicted to be deleterious. rs2062377 was related to a conservative missense variant in TNFRSF11B. On the contrary, rs12821008 was related to the sense variant. Two other SNPs were associated with the 3' untranslated region: rs1346004 in GALNT3 and rs163879 in DCDC5 probably affect splicing regulation. In addition, rs7932354 was predicted to be related to a splice site variant in F2.
Discussion
In this report, we identified that the model proposed in a previous GWAS [18] is available to the Korean population. Furthermore, we found 13 genomic loci significantly associated with BMD variations in the population. Our results reveal that dozens of variants with small effects may contribute to the genetic architecture of BMD and have important roles, regardless of ethnic groups.
Our study supports the important role of several biological pathways related to BMD. The Wnt factor (WNT4) has been known to be associated with BMD in many studies [30,31,32]. A pathway involved in mesenchymal stem cell differentiation (SP7) was also previously known [33, 34]. The RANK-RANKL-OPG pathway (TNFRSF11B) is another pathway influencing BMD [35,36,37]. Interestingly, SNPs in TNFRSF11B and SP7 were associated with BMD-RT, whereas an SNP in WNT4 was associated with BMD-TT. It may suggest differential genetic influences between skeletal sites.
In line with this phenomenon, our results reveal that there is sex- and site-specificity underlying BMD variation. rs13204965 was only significant in women and showed significant sex heterogeneity. Also, in the same manner, the variant was only associated with females in a previous GWAS [18]. In a recent GWAS focusing on women of a specific age (postmenopausal women, age 55-85 years), rs13204965 was associated with BMD at the femoral neck and lumbar spine [13]. In case of rs13245690, a sex-stratified analysis in women showed a more significant result than in pooled samples. These results reflect the sexual dimorphism of bone. Based on the different composition at different skeletal sites, all 12 loci that were significant in pooled samples showed site specificity in BMD. Interestingly, even though the extent of effect size was different from skeletal sites, the direction was consistent between sites. This indicates that BMD-associated loci commonly affect BMD determination across skeletal sites, but the different sensitivity of each site may lead to differential genetic impact on BMD variation.
This could account for the decreased effect size of BMD loci in the Korean population. While the meta-analysis looked at the BMD at the femoral neck and lumbar spine, the KARE focused on BMD at the distal radius and midshaft tibia. Besides, the former were measured by dual-energy X-ray absorptiometry, but the latter were estimated as SOS by quantitative ultrasound. It has been previously shown that BMD of the radius, assessed by ultrasound, poorly coincides with radiologically assessed BMD at the lumbar spine [38]. These represent the limitations of this study- that direct replication at specific sites cannot be performed. Nevertheless, some loci found in the previous GWAS still showed a directionally consistent association in this study. This suggests that a common mechanism underlies BMD throughout different skeletal sites.
In order to identify putative functional variants, SNP-nexus was chosen to annotate proxy SNPs near the BMD-associated loci. In coding regions, some SNPs were expected to present single amino acid substitutions that could be damaging or tolerated. On the other hand, the other SNPs were expected to be synonymous. However, this type of mutation sometimes leads to unsuspected effects. Other annotated SNPs had possible functional effects on the regulation of splicing or the binding activity of transcription factors. To understand how genetic variants affect the phenotype exactly, multi-layered experimental approaches should be carried out in the future.
In conclusion, these findings highlight the polygenic and complex variation of BMD, which could differ even by sex and skeletal site, but provide evidence of the validity in BMD-associated loci between independent populations. This study may contribute to the importance of replication studies in GWASs in understanding the common mechanism underlying BMD.
Acknowledgments
This work was supported by grants from the Korea Centers for Disease Control and Prevention, Republic of Korea (4845-301, 4851-302, 4851-307); the National Research Foundation of Korea (2010-0020259 and 2011-0030049 to TYR); and BK21 PLUS fellowship program funded by the Ministry of Education, Korea (10Z20130-012243 to SH).