Systems Biological Approaches Reveal Non-additive Responses and Multiple Crosstalk Mechanisms between TLR and GPCR Signaling
Article information
Abstract
A variety of ligands differ in their capacity to bind the receptor, elicit gene expression, and modulate physiological responses. Such receptors include Toll-like receptors (TLRs), which recognize various patterns of pathogens and lead to primary innate immune activation against invaders, and G-protein coupled receptors (GPCRs), whose interaction with their cognate ligands activates heterotrimeric G proteins and regulates specific downstream effectors, including immuno-stimulating molecules. Once TLRs are activated, they lead to the expression of hundreds of genes together and bridge the arm of innate and adaptive immune responses. We characterized the gene expression profile of Toll-like receptor 4 (TLR4) in RAW 264.7 cells when it bound with its ligand, 2-keto-3-deoxyoctonate (KDO), the active part of lipopolysaccharide. In addition, to determine the network communications among the TLR, Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and GPCR, we tested RAW 264.7 cells with KDO, interferon-β, or cAMP analog 8-Br. The ligands were also administered as a pair of double and triple combinations.
Introduction
Cells perceive changes in the extracellular environment and create appropriate responses through a complex network of interwoven signaling cascades. For example, lipopolysaccharide (LPS) causes severe inflammatory responses by binding to and signaling through Toll-like receptor 4 (TLR4) on the cell surface, and events downstream of TLR stimulation are extremely dynamic [1, 2]. The inner core regions of all LPS molecules contain at least one 2-keto-3-deoxyoctonic acid (KDO) residue, which possesses endotoxin toxicity that is nearly equivalent to LPS itself [3, 4]. We studied how KDO-mediated signal transduction through TLR4 could be modulated by the presence of interferon-β (IFN-β) and/or 8-bromoadenosine-3',5'-cyclic monophosphate (8-Br). IFN-β uses the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, and 8-Br is a cell-permeable analog of cAMP that is regulated by Gαs- or Gαi-coupled G protein-coupled receptor (GPCR). In response to LPS, TLR4 activates the MyD88-independent pathway to produce tumor necrosis factor α (TNFα) [5], whereas 8-Br activates cyclic AMP response element-binding protein (CREB), which binds to cAMP-responsive element (CRE) sequences, 5'-TGACGTCA-3', and is also a substrate for various cellular kinases [6-10].
The rationale behind the present investigation was to understand the pathway interactions among KDO, IFN-β, and 8-Br and how each of these pathways modulates the signaling from others. If two separate pathways triggered by two distinct ligands regulate the transcription of the same gene, then concomitant stimulation with both of the ligands should result in an additive response equivalent to the sum of the separate responses [11]. In contrast, if the pathways intersect or modulate each other, concomitant addition of the ligands would yield a non-additive response that is either greater than or less than the expected additive response.
Methods
Cells, reagents, and RNA preparation
RAW 264.7 macrophages were stimulated with KDO (100 ng/mL), IFN-β (300 pM), and/or 8-Br (100 µM). These ligands were applied individually or in combinations containing KDO for 60 min and 120 min. Total RNA was extracted using TriPure (Roche, Indianapolis, IN, USA) following the manufacturer's protocol. Each experiment was conducted in duplicate.
Oligonucleotide array fabrication and annotation
Mouse oligonucleotide arrays were fabricated with 15,631 oligomers that were 65 bp or 70 bp long. The oligomers were purchased from Operon (Huntsville, AL, USA) and Sigma-Genosys (St. Louis, MO, USA) and were inkjet-printed onto glass slides by Agilent Technologies (Santa Clara, CA, USA). The list of genes is available through the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) under accession number GPL254.
Gene expression analysis
Hybridization and the resulting analyses were performed as previously described [11-13]. Cy5-labeled cRNA (from ligand-treated cells) and Cy3-labeled cRNA (from time-matched controls) were hybridized in the array. Dye-swap labeling was performed for each pair of samples. The arrays were scanned with an Agilent Scanner G2505A (Agilent Technologies), and image files were extracted with background subtraction and dye normalization using Agilent G2566AA Extraction Software version A.6.1.1 (Agilent Technologies). The microarray data used in this study were deposited into GEO (http://www.ncbi.nlm.nih.gov/geo) under the series accession number GSE11449 and platform ID GPL254.
Quantitative real-time PCR (QRT-PCR)
Selected genes were subjected to real-time PCR analysis in duplicate using the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) following the protocol provided by the manufacturer. The measurement was normalized to a β actin RNA control. The primers of genes are as follows: TNF, F 5'-TCA GCG AGG ACA GCA AGG-3', R 5'-GTG AGT GAA AGG GAC AGA ACC-3'; β-actin, F 5'-CTT TGC AGC TCC TTC GTT GC-3', R 5'-ACG ATG GAG GGG AAT ACAGC-3'. The remaining primer sequences are available upon request.
Cytokine analysis
The Bio-Plex cytokine assay (Bio-Rad Laboratory, Hercules, CA, USA) was used for quantification of cytokines in tissue culture supernatants. In the kit, an antibody directed against each cytokine is covalently coupled to a different color-coded polystyrene bead. The conjugated beads are reacted with a sample containing a known (standard) or unknown amount of cytokines. After unbound cytokines are removed, biotinylated detection antibodies are added to the reaction. The complexes are detected by streptavidin-phycoerythrin, which has fluorescence characteristics. Quantification is carried out by a specialized microtiter plate reader.
Protein phosphorylation
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed to resolve a mixture of proteins by size. The resolved proteins were transferred from the gel to a nitrocellulose membrane (Invitrogen, Carlsbad, CA, USA) via electroelution. The blot was processed for detection of specific phosphoproteins with a mixture of antibodies. All of the incubations and washes were conducted at room temperature with gentle shaking using an oscillating platform shaker. A Molecular Dynamics Storm 860 gel and a blot analysis system (Amersham Pharmacia Biotech, Piscataway, NJ, USA) were used.
Data selection, analysis, and visualization
Features that were differentially expressed in response to ligand stimulation were identified with Linear Models for Microarray Data (LIMMA, http://bioinf.wehi.edu.au/limma). The Benjamini-Hochberg method was used to control the false discovery rate [14]. Four independent experiments were used to identify differentially expressed genes, and p < 0.05 by ANOVA was deemed statistically significant. The average log2-(treated/control) and the average difference (avg.DIF) were hierarchically clustered to visualize the patterns of gene expression changes and the nonadditivity, respectively [15]. Only features with significant expression changes and significant nonadditive responses at one or more time points were included, respectively. Clustering was done one way across the features with experimental conditions aligned in ligand, followed by time course orders. Euclidean correlation coefficient and complete linkage were used as similarity metrics [16]. The hierarchical clustering program that was used was implemented in the Multiple Experiment Viewer (MeV, www.tigr.org/software/tm4/mev.html).
Ingenuity pathway analysis (IPA)
Gene accession numbers, fold-changes of each treatment, and p-value were loaded into the IPA5.0 software (http://www.ingenuity.com). The genes were grouped with their molecular functions and depicted as a network picture, with their direct and indirect relationships represented with the gene ontology by the canonical pathway analysis. All these annotations used Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway maps, and the most significant genes were summarized in the network and as a graph.
Results
General patterns of gene signature in single-ligand studies
We first characterized gene expression profiles after treating RAW 264.7 macrophages with the individual ligands in a kinetic study (60 and 120 min). We found that each of the three ligands, when applied singly, exhibited maximum transcriptional activation after 120 min of stimulation (as analyzed by Limma) (Fig. 1A). We further explored the expression data using k-means/medians clustering (KMC) in MeV, in which we divided the gene expression profiles into clusters in order to identify groups of genes that might participate in similar pathways. The clusters that exhibited changes greater than 2-fold are shown in Fig. 1B. Upon single-ligand treatment, KDO and IFN-β mediated robust transcriptional upregulation, whereas 8-Br stimulation resulted in both up- and downregulation of gene expression. The top-most functions of the genes by all ligand treatment and major networks are given in Fig. 2.
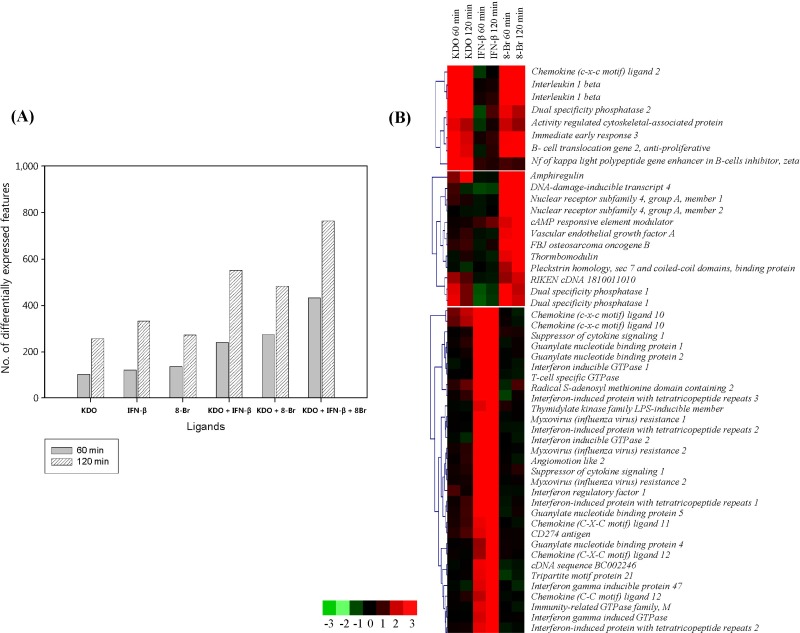
The transcriptional signature of RAW 264.7 cells after stimulation with 2-keto-3-deoxyoctonate (KDO), interferon-β (IFN-β), and 8-bromoadenosine-3',5'-cyclic monophosphate (8-Br). (A) The bars in the diagram represent the numbers of differentially expressed genes at 60 min and 120 min of stimulation, as assessed with Limma. (B) Hierarchical clustering of responses to 60 min and 120 min of stimulation was conducted using K-means clustering and visualized by MeV software. The cluster shown represents those macrophage genes that were significantly regulated by KDO, IFN-β, or 8-Br treatment. Red squares represent genes that were upregulated, black squares indicate no change, and green squares indicate downregulated genes.
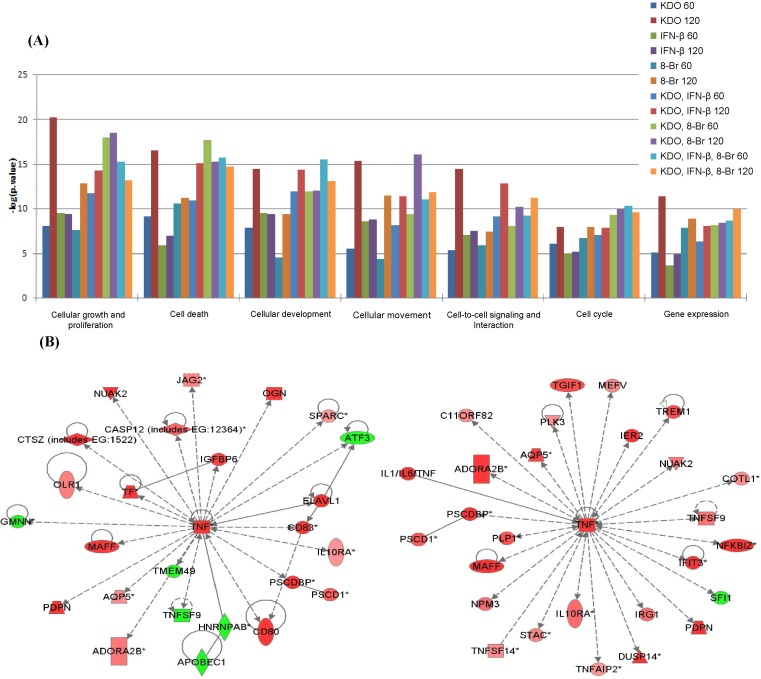
Top functions and tumor necrosis factor (TNF) networks. (A) The top functions for all treatments. As noted, various treatments had various effects on their respective pathways and functions, with 2-keto-3-deoxyoctonate (KDO) the most active ligand, followed by interferon-β (IFN-β) and 8-bromoadenosine-3',5'-cyclic monophosphate (8-Br). (B) TNF networks. Ingenuity pathway analysis (IPA) pictures show the down- and upregulation of the ligand-induced gene expression changes.
KDO regulated activators and repressors of immune signaling
After classifying the genes by KMC, we subcategorized them according to their functions. KDO-regulated genes included transcription factors and genes involved in inflammation, immune responses, signal transduction, cell cycle regulation, cell proliferation, differentiation, metabolism, transport, cell adhesion, and apoptosis (Supplementary Table 1). As expected, KDO-treated cells exhibited a gene regulation pattern similar to that in LPS-treated cells, which included upregulation of Tnf, Il-1α, Il-1β, Cxcl4, and c-Myc. Furthermore, we also observed that KDO regulated several immediate early response genes, such as Egr1, Egr3, Gadd45α, Dusp1, and Dusp2, all of which are reportedly upregulated by LPS [11]. Our results indicated that KDO elicits transcriptional responses that are similar in gene identity and potency to LPS responses.
During the KDO-mediated transcriptional response in RAW 264.7 cells, many of the genes induced at 120 min were candidate feedback repressors of signal transduction [17]. Such repressors were specific to nuclear factor-κB (NF-κB) and Il-1 and include nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor epsilon (Nfκbie, also known as IκBε), nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor zeta (Nfκbiζ, also known as Iκbζ), and interleukin 1 receptor antagonist (Il-1ra). In addition, KDO upregulated Tnfsf9, Tnfaip3, Traf1, and Fas6, indicating that KDO has the potential to elicit inflammation. Its upregulation of Irf1 suggested that KDO also activated interferon-stimulated response elements through the MyD88-independent pathway.
Other KDO-induced genes included IFN-β, CD40, Socs5, Cish, Fosl1, Rgs16, CCR9, Ifit3, and cardiotropin 1 (Ctf1). Among these genes, Socs5 and Cish are feedback inhibitors of the JAK/STAT pathway; both were upregulated after 120 min of stimulation. KDO also promoted Il-10 expression after 120 min of stimulation; this gene contains 2 Creb binding sites in its promoter; so, KDO may have upregulated this gene through enhanced phosphoinositide 3-kinase (PI3K)-v-akt murine thymoma viral oncogene (AKT)-glycogen syntase kinase (GSK) pathway signaling and Creb phosphorylation.
IFN-β induced gene expression of inflammatory/immune responses
Binding of IFN-β to the Type I-IFN receptor activates the JAK/STAT signaling pathway. Phosphorylated Stat proteins regulate gene transcription by binding to conserved sequences within the promoters of IFN-induced genes. IFN-stimulated Stat activation mediates the expression of a second family of transcription factors, the Irf proteins [18]. Irf proteins also mediate transcriptional responses to IFN stimulation. In our study, IFN-β stimulation was associated with Stat2 and Irf1 production, and recent findings indicate that Irf proteins play critical roles in interferon regulation [19].
IFN-β induced a number of genes involved in inflammatory/immune responses, transcription, signaling, and apoptosis. The most highly responsive genes induced by IFN-β were interferon-inducible GTPase 1 (Iigp1), Ifit1, myxovirus (influenza virus) resistance 2 (Mx2), Ifit2, Cxcl9, Cxcl10, Cxcl11, ISG15 ubiquitin-like modifier (ISG15), Socs1, CD40, and TLR3. Secondary responsive genes included Gbp1, Gbp2, Gbp5, Il-13, and Fas. The fold-changes in the expression of these genes are provided in Supplementary Table 2.
8-Br differentially regulated key inflammatory genes
The 8-Br-regulated genes were involved in transcription, signaling, inflammation, apoptosis, metabolism, proteolysis, transport, and cell cycle regulation (Supplementary Table 3). Treating cells with 8-Br upregulated several immediate early transcriptional targets, including nuclear receptor subfamily 4 group A member 2 (Nr4a2), Gadd45 beta, Dusp1, Pde4b, Crem, Cebpb, Vegf, Bag3, and v-maf musculoaponeurotic fibrosarcoma oncogene family protein F (Maff).
Surprisingly, we observed that 8-Br treatment downregulated transcription of Rgs16, Tnf, Cxcl4, Ptger4, Tnfsf9, c-Myc, and Map3k8 by more than 2-fold (Fig. 3A). 8-Br can influence the mitogen-activated protein kianse (MAPK) pathway by downregulating Mapk3k8. Map3k8 activates the kinases p38 and JNK and the transcription factors NF-κB and NF-AT, although its specialized role is to activate the extracellular signal-regulated protein kinase (ERK) pathway downstream of most TLR signals [20-23]. We observed 8-Br-mediated downregulation of 2 proto-oncogenes, c-Myc and Map3k8, and a cell cycle gene, Gadd45α (Supplementary Table 3), which emphasizes that 8-Br cAMP may also have important inhibitory roles in cancer cell proliferation [24, 25].
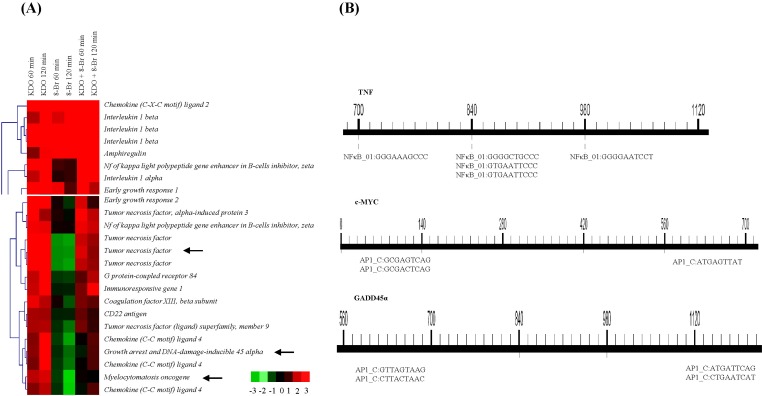
Dual-ligand analysis of 2-keto-3-deoxyoctonate (KDO) and 8-bromoadenosine-3',5'-cyclic monophosphate (8-Br). (A) Clustering of KDO and 8-Br-induced gene expression changes. (B) MotifMogul picture showing the promoter analysis of Tnf, c-Myc, and Gadd45α.
We next hypothesized that the co-downregulated genes may have common transcription factor binding sites; therefore, we examined their promoter regions using TOUCAN (http://homes.esat.kuleuven.be/~saerts/software/toucan.php) and analyzed the genes downregulated by 8-Br using MotifMogul (http://xerad.systemsbiology.net/MotifMogulServer). We found AP1 and/or NF-κB subunit binding sites in the upstream regions of all of the downregulated genes, which adds to evidence from previous studies [26, 27]. Among these genes were several classical targets of NF-κB (for example, Tnf) and AP-1 (for example, c-Myc and Gadd45α); these genes are highlighted with arrows in Fig. 3A and shown in greater detail in Fig. 3B. Since protein kinase A (PKA) activation by cAMP suppresses the MAPK cascade through phosphorylation of Raf [28], we can not exclude the possibility that elevated cAMP may, in part, reduce the transcription of the targeted genes by modulating protein binding to AP-1 sites, by inhibiting the ERK pathway through Raf, and/or by specifically affecting NF-κB subunits. Importantly, Il-1β was up-regulated quickly, whereas Il-1α and Il-10 were not affected prior to 120 min of stimulation. This differential mRNA regulation of genes that are all involved in inflammation suggested that cAMP utilized more than one distinct mechanism to alter the expression of specific sets of genes.
Double-ligand studies: non-additive responses
To understand how the presence of one or more ligands modified the transcriptional responses to the other ligands, we evaluated features that exhibited non-additive responses during the double-ligand studies and grouped them into three categories: significant responses 1) to both ligands; 2) to either ligand A or ligand B; 3) to neither ligand (Table 1). A total of 433 features showed non-additive responses above threshold levels in at least one of the double-ligand studies.
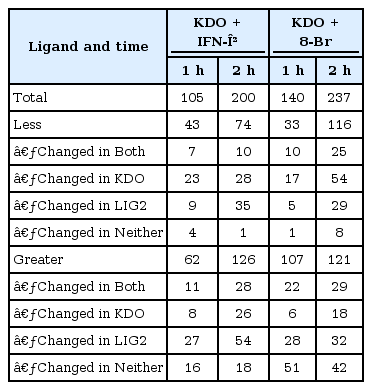
Number of features showing non-additive responses in double-ligand studies with KDO, IFN-β, and 8-Br
In all of the double-ligand studies involving KDO, we found differences in gene transcription that were not present when the ligands were applied individually. Applying KDO together with IFN-β augmented IFN-β-mediated transcriptional responses. In contrast, when KDO was combined with 8-Br, each ligand abrogated the single-ligand response of the other.
KDO enhances IFN-β signaling
KDO enhanced IFN-β-dependent signaling pathways in a time-dependent manner. Since TLR4 activation can also produce IFN-β, the KDO-mediated enhancement suggested cooperation between exogenously applied and endogenously produced IFN-β (Supplementary Table 4). Several IFN-β and KDO primary responsive genes were upregulated, including genes that activate the immune system. In some cases, the IFN-β gene signature dominated over the KDO gene signature. For example, IFN-β inhibited two KDO primary responsive genes, Il-1ra and Nfκbζ, but KDO did not change the expression of any of the interferon-responsive primary genes. The expression levels of select genes were validated by using QRT-PCR (Fig. 4).
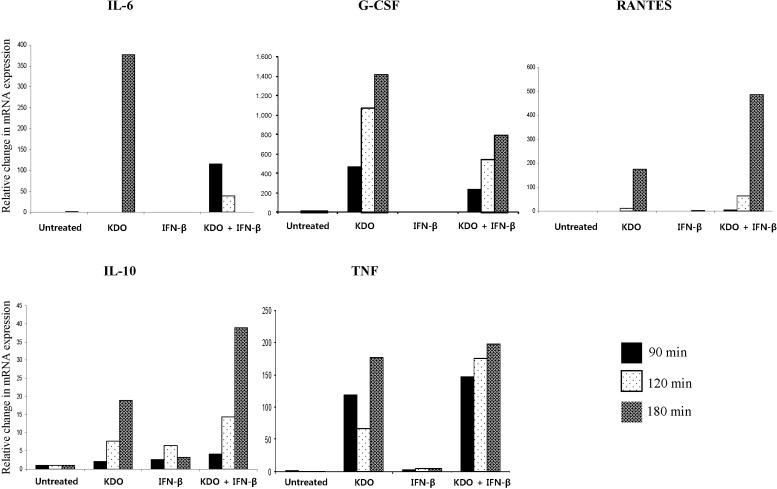
Quantitative real-time PCR validation of genes regulated by 2-keto-3-deoxyoctonate (KDO), interferon-β (IFN-β), or KDO + IFN-β. KDO enhanced the expression of RANTES and interleukin 10 (IL-10) together with IFN-β. G-CSF, granulocyte-colony stimulating factor; TNF, tumor necrosis factor.
Cytokine assays, conducted following a combination of LPS and IFN-β treatment, also demonstrated non-additive enhancement of Il-6, Il-10, RANTES, and TNF protein levels (Fig. 5A). This increase could be due to synergistic production of transcription factors in response to these ligands, apart from their classical transcription factors. It is likely that the rapid, greater-than-2-fold increase in Socs1 and Cish production may have contributed to the attenuated expression of the KDO secondary responsive genes [29, 30]. Il-10 is an anti-inflammatory cytokine regulated through the PI3K-AKT-GSK-CREB pathway [31, 32], and we noted its expression after 120 min of stimulation, suggesting that the PI3K pathway may have been activated after 60 min of stimulation.
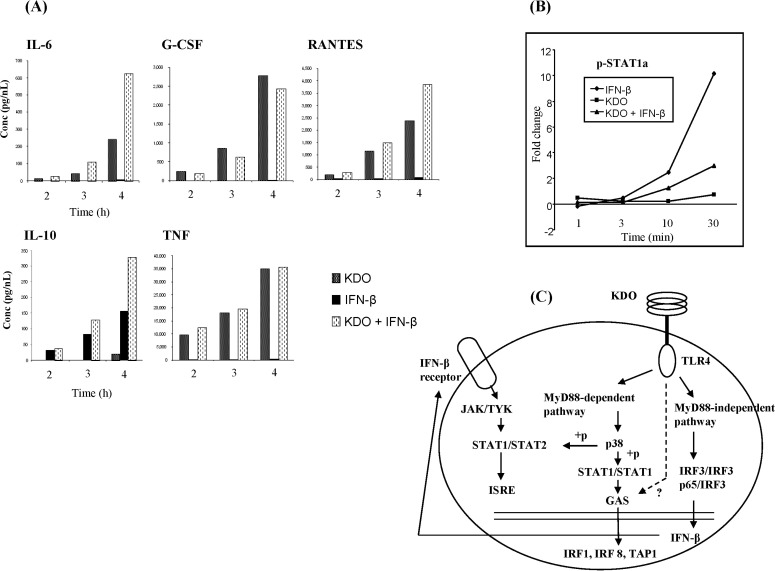
Dual-ligand analysis of 2-keto-3-deoxyoctonate (KDO) and interferon-β (IFN-β). (A) Cytokine analysis of non-additive genes in KDO and IFN-β stimulation. (B) Stat1 phosphoprotein analysis upon KDO and IFN-β stimulation. (C) KDO and IFN-β signaling mechanisms. GAS, gamma activated sequence; G-CSF, granulocyte-colony stimulating factor; IL, interleukin; IRF3, interferon-regulatory factor 3; ISRE, interferon-stimulated response element; JAK/TYK, Janus kinase/tyrosine kinase; STAT, signal transducer and activator of transcription; TLR4, Toll-like receptor 4; TNF, tumor necrosis factor.
KDO- and IFN-β-treated cells exhibited upregulation of many interferon-regulatory factors (IRFs), including Irf1, Irf4, and Irf8. Irf4 inhibits the expression of MyD88-dependent IRF genes by competing with IRF5 for binding to the same region of MyD88 [33]. Combined KDO and IFN treatment also upregulated genes with IRF3 binding sites in their promoters; i.e., Ifit1, Ifit2, Ifit3, Isg15, and Gbp1. Our gene profiling results suggested that IRF3 is sufficient to induce antiviral responses at the initial activation time point, consistent with an earlier report that demonstrated a role for IRF3-dependent genes in establishing antiviral responses in Jurkat cells [34].
KDO and IFN-β regulate gamma activated sequence (GAS) genes together
Interestingly, two of the genes that exhibited non-additive responses are produced only by IFN-γ GAS elements: Irf8, previously known as interferon consensus sequence-binding protein 1 (ICSBP), and transporter associated with antigen processing-1 (Tap1). Upregulation of GAS-driven genes requires the Stat1 homodimer exclusively but not NF-κB; they are both produced by IFN-γ. Interestingly, in our study, we did not observe elevated expression of IFN-γ or STAT1 mRNA, but we still observed the production of GAS-driven genes.
Two phosphorylation sites, Tyr-701 and Ser-727, differentially regulate Stat1, and phosphorylation of both sites is required for maximal transcriptional activity [35, 36]. A previous study demonstrated that Stat1 is phosphorylated on both Tyr-701 and Ser-727 in LPS-treated cells after 2 h [37]. We surmise that this enhanced phosphorylation of STAT1 may be mediated by KDO and IFN-β. However, when we conducted STAT1 phosphoprotein measurements with KDO and IFN-β in combination, we observed a 3-fold upregulation in phosphorylated STAT1 after 30 min (Fig. 5B). This increase in phosphorylated STAT1 during double-ligand stimulation was lower than the increase in phosphorylation of STAT1 induced by IFN-β alone (10-fold). Our previous studies with LPS- and IFN-γ-treated RAW 264.7 cells also showed non-additive increases in Irf8 and Tap1 gene expression [11]. This elevated GAS gene expression definitively indicated that KDO and IFN-β activate GAS promoters, either directly or indirectly.
KDO abrogated the anti-inflammatory properties of 8-Br
Supplementary Table 5 shows the genes regulated upon double-ligand treatment with KDO and 8-Br. Importantly, combined KDO and 8-Br application increased the transcription of Tnf but did not affect Ccl4, Map3k8, or c-Myc (Fig. 3A). Since 8-Br downregulated these genes during the single-ligand application studies, these results suggested that the KDO-induced pathway attenuated these 8-Br-induced changes. We classified the direct targets of CREB (major TF induced by cAMP analogs) and NF-κB (direct target of KDO) to see which of the major transcription factors are affected (Table 2). For CREB, we searched in the website developed by Marc Montminy, which takes advantage of CHIP-on-chip data (http://natural.salk.edu/CREB). For potent NF-κB sites, we looked in the website http://www.bu.edu/nf-kb. Surprisingly, both of these transcription factors are not affected by double ligand treatment of KDO and 8-Br, reinsisting that KDO directly inhibits 8-Br-induced changes. CREB can be induced to a certain extent through the ERK pathway by KDO; however, such a claim remains to be proven in the future studies. This approach further raises a question on by which mechanism, if not CREB and NF-κB, KDO attenuates 8-Br induced changes, which also remains to be tested. One possible explanation for this phenomenon is that KDO stimulation rapidly downregulated the 8-Br-induced increase in intracellular cAMP through the action of phosphodiesterases (PDEs) that transform cAMP into inactive AMP [38]. In support of this idea, LPS stimulation of mouse peripheral leukocytes induced Pde4b mRNA accumulation and increased PDE4 activity [39]. This increase in PDE4 activity leads to a decrease in cAMP, thus removing the cAMP constraint and allowing for full induction of Tnf mRNA. PKA phosphorylation inhibits Raf1 directly [40, 41], and this inhibition occurs downstream of Ras [42, 43] to inhibit Ras-dependent signaling to ERK [44]. Hence, Raf can be the crosstalk point between the KDO and 8-Br signal transduction pathways.
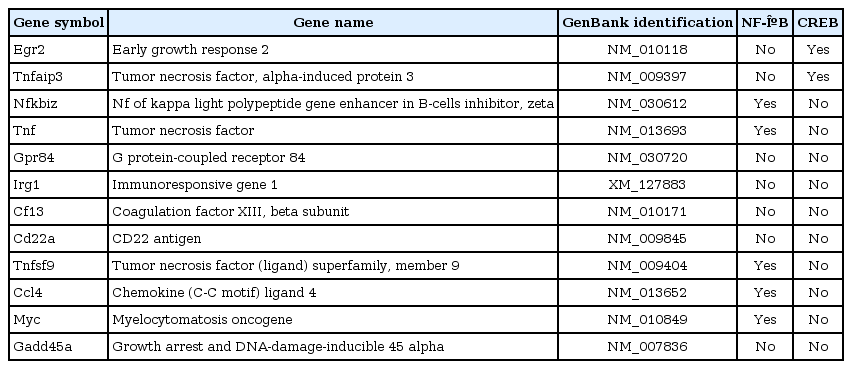
The potent CREB and NF-κB sites were searched, and their presence or absence is listed
KDO and IFN-β suppressed the effects of 8-Br in triple-ligand studies
Supplementary Table 6 shows the transcriptional responses in RAW 264.7 cells upon triple-ligand stimulation. We observed the following effects: 1) upregulation of the primary and secondary KDO- and IFN-β-responsive genes; 2) production of various inflammatory cytokines and their receptors (Il-1a, Il-1b, Il-6, Il-20, Il-17rb, Il-15, Il-18, Il-13, Il-4ra, Il-10ra, and Tnf) and the anti-inflammatory cytokine Il-10; 3) induction of apoptosis-inducing transcription factors (Fosl-1, Atf3, Egr1, Egr2, Fos-b, Jun dimerization protein [Jdp], and Irf14); and 4) up-regulation of multiple G-protein-coupled receptors, like leukotriene B4 receptor1 (Ltb4r1), complement component 5a receptor 1 (C5ar1), chemokine (c-c) receptor 1 (Ccr1), Gpr132, Gpr65, and calcitonin receptor-like (Calcr1). In these triple-ligand studies, the transcription factors exerting the greatest regulatory control over gene expression were NF-κB, Ap1, Nfat, Stat2, and Stat3. Our data demonstrated that concomitant stimulation with all three ligands induced KDO and IFN-β primary and secondary responsive genes, but it repressed 8-Br-induced transcriptional changes.
Triple-ligand stimulation revealed Il-6 gene induction does not require de novo synthesis of NF-κBζ
We observed an interesting transcriptional change in gene clusters (Fig. 6). NF-κBζ is weakly expressed in cells but induced to a marked extent upon LPS stimulation and regulates a set of gene expression, including Il-6 [45]. However, a recent study demonstrated that Il-6 promoter can also be activated by unknown molecules acting far from the transcriptional active site by cis-regulatory molecules [46]. We observed the same phenomenon when we stimulated RAW 264.7 cells with KDO. NF-κBζ was upregulated by KDO at 60 min, and Il-6 was not induced by KDO till 120 min, which is in line with a previous observation [45]. This observation is very novel to be explained. Triple-ligand studies showed upregulation of Il-6 as early as 60 min (about 6-fold). This indicates that though KDO-induced pathways need de novo synthesis of NF-κBζ for Il-6 production, it does not require de novo synthesis of NF-κBζ for the induction of Il-6 when combined with IFN-β and 8-Br. We then looked for the possible ligand that could have mediated this and found that KDO and 8-Br never produced Il-6 on single-ligand stimulation. In that case, the only remaining candidate is IFN-β. IFN-β upregulated Il-6 in our experiment at 120 min (TLR3 can produce Il-6 due to the late phase of NF-κB activation). IFN-β also increased mRNAs of Atf3, Ccl4, and Map3k8 at 120 min. It is of interest to know what pathway regulates these genes in future studies, since most of them were reported to be regulated by a MyD88-dependent pathway during TLR signaling. Either the presence of a putative STAT1 site is sufficient to induce the MyD88-dependent genes or IFN-β might have crosstalk with the MAPK pathway.
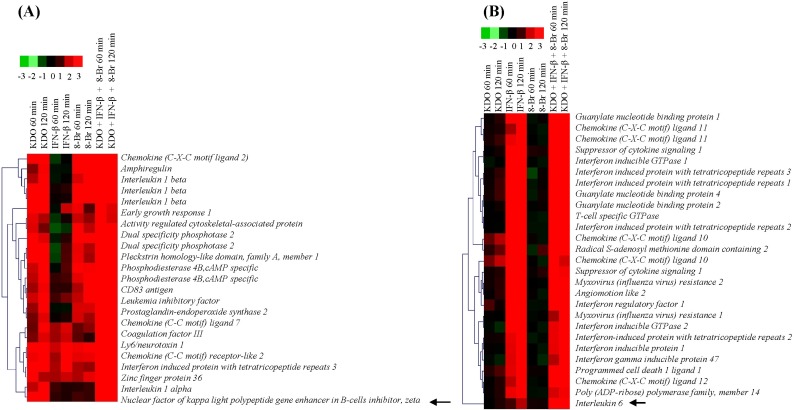
Regulation of NF-κBζ and interleukin 6 (Il-6) by all ligands. (A) NF-κBζ, which is highlighted in black arrow, was regulated by 2-keto-3-deoxyoctonate (KDO) as well as by triple-ligand stimulation in RAW 264.7 cells. (B) Il-6 was regulated by interferon-β (IFN-β) at 120 min, but triple ligands upregulated Il-6 robustly as early as 60 min itself. 8-Br, 8-bromoadenosine-3',5'-cyclic monophosphate.
Discussion
In RAW cells and primary macrophages, LPS-induced signaling is mainly passed through the activation of the NF-κB/IκB kinase (IKK) pathways. The p50 and RelA complex mediates the majority of genes induced by LPS. In our studies, many induced genes appeared to be activated as a primary consequence of initial NF-κB activation. Although the induction of the genes was through the NF-κB pathway, few genes were differentially expressed when we combined the ligands. We have restricted our studies to 2 h; hence, we can not emphasize on the secondary sequences of NF-κB, but we observed that most of the induced genes have putative or known AP-1 or IRF-3 sites. The gene expression in dual treatment of IFN-β and KDO was specifically mediated by STAT proteins and NF-κB. Also, gene transcription during macrophage activation through TLR4 and the elevation of IFN-β simultaneously resulted in alterations in the chromatin structure of gene promoters that further influenced transcription initiation events.
Combining IFN-β and KDO induced the IFN-β secondary responsive genes Gbp1, Gbp2, and Gbp5 by more than 4-fold, but it attenuated several KDO secondary responsive genes, including Socs5, Cish, Fosl, Rgs16, Cxcr9, Ifit3, and Ctf1. In contrast, dual KDO and IFN-β treatment enhanced the expression of several transcription factors after 60 and 120 min of stimulation, most notably Atf3. Atf3 is a stress-inducible member of the Atf/Creb transcription factor family [47], and according to a recent systems biological approach, Atf3 negatively regulates TLR4-induced Il-6 and Il-12 transcription [48]. As evident from our QRT-PCR results, IFN-β never regulated MyD88-independent genes except Il-10. However, IFN-β treatment during TLR4 signaling increased Rantes, Il-10, and Tnf levels. It is interesting to note that that there should be a competition for the common signaling repressors and activators of the inflammatory ligand TNF and anti-inflammatory ligand Il-10 at approximately 4 h of signaling. It is also worthwhile to note that if TNF pathways dominate, it should lead to increased Il-10-repressed genes and direct the cells into the apoptosis pathway.
In addition to this effect, we observed upregulation but not enhanced levels of NF-κB p105, implicating this subunit in KDO and IFN-β responses not only through IκB-NF-κB interaction but also through transcriptional regulation. The elevated KDO and IFN-β primary and secondary responsive genes were not unexpected, considering that we observed higher NF-κB signaling and endogenous production of IFN-β under double-ligand stimulation. This suggests that there exists some crosstalk and pathway modulation in a positive way that increased the levels of gene expression.
Macrophages, when infected with pathogenic Gram-negative bacteria, lead to the induction of about 1,000 genes, and almost all of these genes were reported to be induced only though the TLR4 signaling cascade. Excessive production of cytokines or inflammatory agents is always associated with sepsis and shock. The data presented in our microarray also show that KDO and LPS induce identical gene expression profiles. This suggests that the contribution of other family members of TLR is minor, and TLR4 on the whole contributes to KDO-induced gene expression changes. Further, most of these gene responses were mediated by the MyD88-dependent pathway, which is essential for the optimal induction of many well-established cytokines, like Il-1, TNF, Il-10, Il-12, and Il-6. On the other hand, in our microarray studies, we observed that Tnf and other inflammatory genes were downregulated by 8-Br, whereas upregulated Il-10 levels. However, in double-ligand treatment, KDO abrogated all the gene responses of 8-Br. cAMP-dependent and -independent signaling mechanisms may be involved in mediating the gene regulation during double-ligand stimulation with KDO and 8-Br.
Another observation was the differential regulation of mRNAs of Il-1β, Il-1α, and Tnf by KDO and 8-Br. Ccl3, Ccl4, Il-1β, Tnf-α, and Gadd45β are the genes whose promoters have been proven to be regulated by RelA [49]. In our studies, all these genes were found to be upregulated at 60 min of KDO treatment, which is consistent with a previous report [50]. However, 8-Br increased Il-1β levels, but it did not increase Tnf levels. Both genes are regulated through the MyD88-dependent signaling. On the other hand, there was no change in Il-1α levels. In double-ligand stimulation, all these changes were reversed. We tried to find answers through the promoter and transcription factor search analysis. Transcription factors evolved to regulate the gene expression, and they have the ability to modify the expression of a given gene. Since Tnf is downregulated by 8-Br at 60 min itself, it should be happening either at the promoter level or at the primary pathway level. Although Tnf has AP1 binding sites, it is not sufficient to induce Tnf mRNA, whereas NF-κB is necessary for its upregulation. 8-Br should have affected the translocation of p65/p50 subunit to the nucleus.
Il-1β is highly expressed in activated macrophages and monocytes, and LPS is a well-established stimulus for its induction. Il-1β has one or more CREB binding sites in the promoter. Since 8-Br activates CREB, the presence of one or more CREB sites seems to be sufficient for its induction. On the other hand, Il-1α mRNA level was not changed. With the information available on its regulation, it seems that the AP1 site is necessary for its induction. 8-Br-induced pathways may have either directly or indirectly affected AP1 translocation to the nucleus. In this context, to support our notion, we observed the downregulation of some AP1 genes. 8-Br-induced pathways modulated the MAPK pathway, which must have happened as a dual channel. First, 8-Br may have inhibited the pathway upstream of AP1. Second, it should have inhibited the translocation of the specific subunits of AP1 into the nucleus. Gene regulation studies of 8-Br suggest the following mechanism of the pathway activation/repression: 1) 8-Br affected NF-κB activity directly; 2) 8-Br-induced CREB is sufficient to induce Il-1β expression; and 3) 8-Br affected the different subunits of AP1 specifically, and at the same time, it inhibited the pathway of MAPK upstream of AP1.
PDEs are enzymes that catalyze the hydrolysis of cAMP and cGMP, inactivating these second messengers [51]. PDE consists of 11 families, and amongst them four, including PDE4, PDE7, and PDE8, are cAMP-specific [52]. PDE4 isoforms are major inactivators of cAMP signaling [53]. In our studies, stimulation of TLR caused a major upregulation of PDE4B but not the paralogs PDE4A or PDE4D. Ablation of PDE4B impacted LPS signaling, and TNF-α mRNA and protein were decreased by >50% in PDE4B-/- but not in PDE4A-/- or PDE4D-/- macrophages [54]. This indicates the powerful role of PDE4B in macrophages, by inhibiting cAMP and its critical role in LPS signaling. Another possible explanation for this pathway modulation is the interaction between NF-κB and PKA. Since the catalytic subunit of PKA works as a complex with NF-κB/IκB in the inactive state [55], stimuli that degrade IκB also activate the associated PKA catalytic subunit, which promotes the phosphorylation of p65 at Ser 276 and increases the transcriptional activity of NF-κB. Two GPCR ligands, isoproterenol and prostaglandin E2, induce cAMP through the activation of Gαs and adenylate cyclase. cAMP production inhibits pro-inflammatory mediators, including TNF and nitric oxide in RAW 264.7 cells [56, 57]. Apart from this, multiple signaling pathways or mechanisms are likely to be involved in mediating gene responses to KDO and 8-Br. In some cases, an increase in cAMP level by pituitary adenylate cyclase blocks IκB degradation, p65/Rel translocation, and/or the rearrangement of CREB/Jun/CREB-binding protein (CBP) complexes [58]. In other cases, an increase in cAMP is directly proportional to the decrease in MAPK/ERK kinase (MEK) kinase pathways [59], and transactivating properties of PKA are reported to be involved in NF-κB activation [60].
After 60 min, triple-ligand treatment upregulated many transcription factors, such as Nr4a2, Crem, Atf3, Irf8, Nfkbp100, Egr2, Egr1, Cish, Stat3, Stat2, Irf1, and FosB. After 120 min of stimulation, we observed upregulated zinc finger and BTB domain-containing 32 (Zbtb32), Fosl1, RelB, Irf7, LPS-induced TNF-α factor (Litaf), and Jundm2. Although the regulatory mechanisms of these genes are complex, they all culminate in the production of various pro-inflammatory cytokines. NIH3T3 cells, when stimulated with the cAMP analog forskolin, activate p38 by PKA, which in turn phosphorylates CREB through RSK2. However, this process is delayed in comparison with the direct phosphorylation of CREB by PKA [61]. This study shows that there exist crosstalk mechanisms between PKA and p38, thus allowing the cells to prolong the incoming signal. Both LPS and the cAMP analog 8-Br activate p38, like forskolin, forming the crosstalk points between GPCR and Toll-like receptor. This mechanism would have partly contributed to the observed dominant gene signature of KDO in triple-ligand studies. We also noted that there exist substantial differences in the gene expression profiles of RAW 264.7 cells and primary macrophages. For example, TNF alpha is produced highly in RAW 264.7 cells than in macrophages, and there are quantitative and temporal differences between these cells.
The KDO-induced gene signature dominated over IFN-β and the 8-Br-induced gene signature. In addition, we also explained the genes that have the ability to act as positive (Tnf, Il-1) [62, 63] and negative feedback (Il-1ra) [64] regulators of KDO signaling. Excessive activation of cytokines is associated with septic shock and clinical consequences of inflammatory disorders. It is also of interest that many genes induced by 8-Br have the potential to act as anti-inflammatory actions. Thus, our studies prove that elevating intracellular cAMP levels during the times of immunological stress might ameliorate the shock-induced mortality.
Taken together, our study identified several target genes that are activated/repressed by KDO, IFN-β, and/or 8-Br in RAW 264.7 cells. Based on these data, we also proposed regulatory signaling networks that could be responsible for the crosstalk between Toll-like receptors and non-Toll-like receptors, such as GPCRs. These findings not only highlight the decisive regulation of various genes by three important ligands but also add depth to our broad understanding of biological signaling connections between complex systems of functionally interacting macromolecules. Gene responses in macrophages to LPS vary between different methylation patterns [65], sub-clones of RAW 264.7 cells [66], chromatin structure of gene promoters, and chromatin remodeling events [67]. To define the action of a given ligand, it is necessary to rule out these options and particularly emphasize on the true effects of a ligand. Though our studies form the basis for the novel regulatory pathways of gene induction during macrophage action, future studies are warranted to construct a signaling map.
Acknowledgments
This work was supported by the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (2012016803). This work was also partly supported by the Korea Food & Drug Administration (10182KFDA992-2302) and the Priority Research Centers Program (NRF 2010-0028294).
References
Supplementary materials
Supplementary data including six tables can be found with this article online at http://www.genominfo.org/src/sm/gni-10-153-s001.pdf.