Current Challenges in Bacterial Transcriptomics
Article information
Abstract
Over the past decade or so, dramatic developments in our ability to experimentally determine the content and function of genomes have taken place. In particular, next-generation sequencing technologies are now inspiring a new understanding of bacterial transcriptomes on a global scale. In bacterial cells, whole-transcriptome studies have not received attention, owing to the general view that bacterial genomes are simple. However, several recent RNA sequencing results are revealing unexpected levels of complexity in bacterial transcriptomes, indicating that the transcribed regions of genomes are much larger and complex than previously anticipated. In particular, these data show a wide array of small RNAs, antisense RNAs, and alternative transcripts. Here, we review how current transcriptomics are now revolutionizing our understanding of the complexity and regulation of bacterial transcriptomes.
Introduction
An operon, which is an independent unit of biochemical function in the bacterial genome, consists of functionally related genes under the control of a single promoter [1]. Thus, the cluster of genes in an operon is transcribed into a single mRNA molecule, referred to as a polycistronic system. This foundational concept of bacterial genome organization has been leading to the discovery of fundamental regulatory mechanisms of bacterial transcription. Bacterial transcription is a biological process that is initiated from a promoter by the RNA polymerase (RNAP) holoenzyme complex, consisting of a single RNAP core unit and a σ-factor subunit. Association with different σ-factor subunits provides promoter specificity to the RNAP core unit. Seven species of σ-factor subunits, for example, have been identified in Escherichia coli, and each participates in the transcription of a specific set of genes in response to growth conditions [2]. In addition to the σ-factor association, transcription is either activated or repressed by a wide range of transcription factors, such as the Lac repressor of the lac operon. It was estimated that E. coli encodes roughly 300 regulatory DNA-binding proteins, of which 35% are activators, 43% are repressors, and 22% are dual regulators [3].
Bacterial gene expression can be regulated by other regulatory factors, in addition to regulatory DNA-binding proteins, such as small regulatory RNAs and internal promoters within operons, which increase the transcriptome complexity [4, 5]. Over the last decade or so, many of these examples have been discovered by quantitative analysis of bacterial transcriptomes, which are defined as the complete set of cellular RNA transcripts. Although DNA microarrays have provided comprehensive information on the transcriptome's complexity in bacterial cells [6, 7], the advent of next-generation sequencing (NGS) has dramatically accelerated our analytical capacity via high-throughput transcriptome sequencing (RNA-seq) in combination with mRNA enrichment methods [8]. This large amount of data has revealed many unexpected features in the bacterial transcriptome, including gene structures, multiple promoters, and RNA-based regulation [7, 9, 10]. In some cases, small RNAs (sRNAs) account for 10% to 20% of bacterial RNA products, which have important regulatory roles [8]. These new findings suggest that bacterial transcription is much more complicated and subtle than previously thought. In this review, we discuss advances in bacterial transcriptomics, based on RNA-seq, and the current understanding of the complexity and regulation of bacterial transcriptomes.
Determination of Bacterial Transcriptomes
Like eukaryotes, bacterial transcriptome analyses have been initiated by the development of microarrays [11]. In particular, high-density tiling arrays, which consist of hundreds of thousands of DNA oligonucleotide probes representing both strands of a given genome, have been massively used to study bacterial transcriptomes. The major advantage of tiling arrays is to interrogate the boundaries of the entire set of transcripts in a cell without taking account of genome annotation [12]. Consequently, this technical advance discovered many new RNA molecules, such as regulatory sRNAs. Based on this experimental approach, comprehensive transcriptomics have recently been published for Bacillus subtilis [13], Halobacterium salinarum [14], Mycoplasma pneumonia [6], Caulobacter crescentus [15], Listeria monocytogenes [16], and E. coli [10]. One of these studies, for example, focused on the analysis of transcriptionally active regions across an entire genome of Listeria monocytogenes, revealing the complete Listeria operon map as well as various types of RNAs, including at least two of which are involved in virulence in mice [16]. Although the high-density tiling arrays were able to interrogate the bacterial transcriptomes without genome annotation, the array-based approach is limited by a high rate of noise due to cross-hybridization, the low dynamic range of detection due to signal saturation, and the inability to detect low copy number of transcripts [17].
High-throughput sequencing of RNAs using NGS methods has overcome some drawbacks of the array-based approach. Currently, commercially available NGS systems include the Roche 454 system, based on the pyrosequencing; the Illumina sequencing system, based on sequencing by synthesis; the SOLiD system, based on sequencing by oligonucleotide ligation and detection; and the Ion torrent system, based on the use of a semiconductor-based sequencing technique [18, 19]. Also, third-generation sequencing technologies have been actively developed to determine the sequence directly from a single DNA molecule without DNA library amplification [18]. Over the conventional Sanger sequencing method, the primary advantage offered by NGS methods is the inexpensive production of large volumes of sequence data, which can be used to identify and quantify rare transcripts without prior knowledge of a particular gene [19, 20].
Unlike a hybridization-based array approach, RNA-seq allows unambiguous mapping of transcripts to unique regions of the genome with single-base resolution; hence, there is essentially lower background noise [21]. In addition to the accurate quantification of a transcriptome consisting of known genes, RNA-seq allows researchers to determine the correct gene annotation, expressed single-nucleotide polymorphisms, novel genes, and RNAs with high levels of reproducibility for both technical and biological replicates. However, bacterial mRNA can exist as little as 1% to 5% of total RNA; so, mRNA enrichment is a challenging step to obtain sufficient transcript coverage [8]. Currently, several methods are being used to remove the rRNA and tRNA fraction from the total RNA pool. Among those methods, terminator 5'-phosphate-dependent exonuclease treatment has been successfully applied to enrich primary transcriptome by reducing processed or degraded RNAs with a 5'-monophosphate end (e.g., rRNAs and tRNAs) [9, 10]. To remove the rRNA fraction from the total RNA pool, Ribo-Zero rRNA removal kits, using biotinylated probes that selectively bind rRNA, have been used successfully for a wide range of organisms, from bacteria to human (Epicentre).
The requirements of the ideal RNA-seq method are strand-specificity and quantitation across a wide dynamic range [22, 23]. However, a common strategy of an RNA-seq library preparation is to convert single-stranded RNA molecules into double-stranded cDNA fragments of certain sizes, flanked by platform-specific adapter sequences. This means that double-strand cDNA is synthesized from randomly primed hexamers, which cannot provide information on the transcribed strands. A major shortcoming of conventional RNA-seq methods is therefore the lack of RNA polarity information. The polarity of RNA transcripts is very useful in facilitating subsequent computational analyses, including the correction of novel gene annotations and the detection of overlapping genetic features encoded in opposite orientations. In particular, polarity information is important for understanding small bacterial genomes, in which genes are densely coded, with overlapping untranslated regions (UTRs) or open reading frames (ORFs) [23].
However, the recently developed strand-specific RNA-seq analysis enhances the value of bacterial transcriptomics. In terms of how to mark the transcribed strand, several methods can be categorized largely into two approaches. One is marking a strand by orientation-dependent adaptor ligation to the 5' and 3' ends of the RNA transcript, such as RNA ligation [24], SMART [25], NNSR priming [26], and SMART-RNA ligation [23]. The other is marking one strand by chemical modification, either on the RNA itself by bisulfite treatment [27] or on the second strand cDNA by dUTP incorporation [28]. Among several strand-specific RNA-seq methods, the dUTP method was identified as the leading protocol because of its wide range of coverage and availability of paired-end sequencing (Fig. 1A) [23]. Briefly, the dUTP method starts with converting the initial mRNA into cDNA. This first cDNA strand is used as a template for second cDNA strand synthesis, which incorporates dUTP instead of dTTP. After marking both ends of the cDNA by ligation of adaptor sequences, the second strand is subsequently degraded by uracil-N-glycosylase treatment, which selectively removes the uracil site on the DNA. Finally, the remaining first-strand cDNA, with directional adaptor sequences, can be sequenced on the Illumina Platform using both single-read and paired-end sequencing [28].
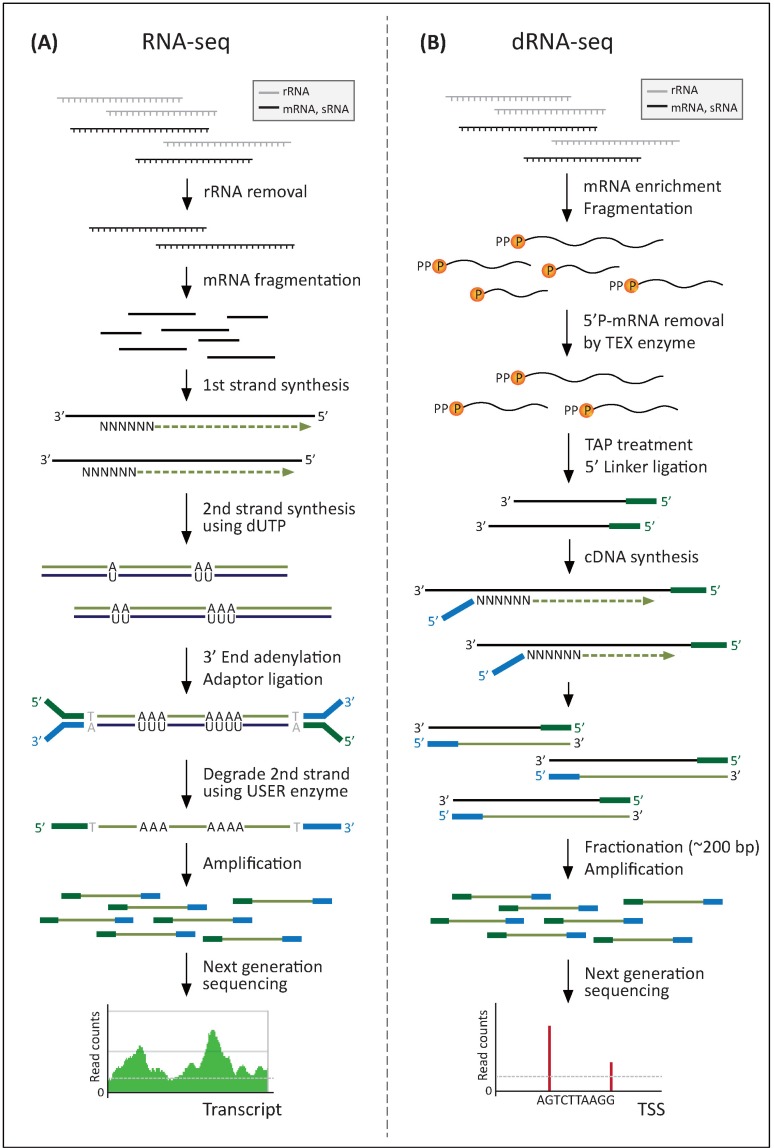
Methods for strand-specific RNA-seq. (A) RNA-seq method using dUTP incorporation and uracil-N-glycosylase treatment. (B) dRNA-seq method using 5'-monophosphate (5'P)-dependent terminator exonuclease (TEX) and terminal tobacco acid pyrophosphatase (TAP). TSS, transcription start site.
With these technological breakthroughs, several bacterial RNA-seq studies showed that bacterial transcription is not as simple as previously thought [10, 29-32]. For example, in the transcriptomic analyses of Salmonella typhimurium, E. coli, Vibrio cholera, and Xanthomonas campestris pv. vesicatoria, it was revealed that noncoding RNAs play an important role in diverse regulatory mechanisms [29-32]. In addition, the complexity of the E. coli transcription unit indicates that bacterial transcription is regulated by the differential use of diverse transcription start sites (TSSs), with corresponding transcription factors under given environmental conditions [10].
Primary Transcriptome Analysis
In bacteria, cellular RNA comprises primary transcripts that are marked by a 5'-triphosphate (5'PPP) group and processed or degraded RNAs that carry either a 5'-monophosphate (5'P) or 5'-hydroxyl group (5'OH) [7, 9]. In particular, primary transcriptome analysis is beneficial for improving the annotation of any sequenced bacterial genome. Due to the absence of a poly(A) tail at the 3'-end and the instability of mRNAs with very short half-lives, however, bacterial primary transcriptome analysis has been challenging [8]. Also, cellular RNA was considered to consist mainly of rRNA and tRNA in prokaryotic cells; therefore, mRNA enrichment is essential to determine the primary transcriptome from RNA-seq. Differential RNA-seq (dRNA-seq) enriches RNA samples for primary transcripts by use of a 5'P-dependent terminator exonuclease (TEX) that specifically degrades RNAs carrying a 5'P, leading to relative enrichment of primary transcripts and depletion of processed RNAs. Then, the 5'-end positions of the primary transcripts can be identified by comparison of cDNA libraries generated with and without TEX treatment (Fig. 1B) [9, 31, 33-36]. The 5'-end of RNA transcripts, corresponding to the TSS, is mostly related with the regulation of transcribed genes, including promoters, ribosome binding sites, transcription factor binding sites, antisense RNAs (asRNAs), and other regulatory noncoding RNAs (ncRNAs).
Transcription Start Site
Bacterial TSSs have been identified mainly through primer extension and S1 nuclease protection mapping assay, but these methods are labor-intensive (Fig. 2A) [37]. High-density tiling arrays have also been used for large-scale TSS mapping in E. coli and Caulobacter crescentus, with limited mapping resolution [15, 38]. In addition, indirect TSS identification in the E. coli transcriptome has been made by RNAP chromatin immunoprecipitation, coupled with microarray (ChIP-chip) [39]. More recently, dRNA-seq has been developed to map TSSs in Helicobacter pylori [9]. It revealed 1907 TSSs, which were found in upstream regions of annotated mRNAs within annotated genes on the same DNA strand or on the opposite DNA strand and in regions without any annotated genes in proximity. In addition to the 337 primary operons defined by the TSSs, it obtained 126 additional suboperons and 66 monocistrons overlapping the 3' part of polycistrons in H. pylori. Alternative TSSs for the determination of suboperons (or transcription units) have been determined through the integration of massive-scale sequencing and 5'-rapid amplification of cDNA ends (5'-RACE) method in E. coli (Fig. 2B) [10]. The consecutive iterations of experiments identified a total of 4,133 TSSs. Interestingly, it was found that ~35% of promoters contained multiple TSSs, defining alternative transcription units in the E. coli genome. For example, the transcription factor Lrp activates stpA, encoding an H-NS-like DNA-binding protein from a dominant TSS position (2,796,558). However, two other TSSs are used less in the exponential growth phase. These results show that the bacterial transcriptome is actively regulated by means of alternative TSSs in response to differential environmental conditions. dRNA-seq method has been applied to detect genomewide TSS maps in other bacterial species. Interestingly, the cyanobacterium Synechocystis contains 3,537 TSSs, 64% of which were assigned to asRNAs or ncRNAs [40]. In conclusion, the dRNA-seq approach is a strong tool for annotation of operon structures, transcription units (or suboperons), and detection of TSSs, providing important insights into the understanding of a comprehensive transcription regulatory network.
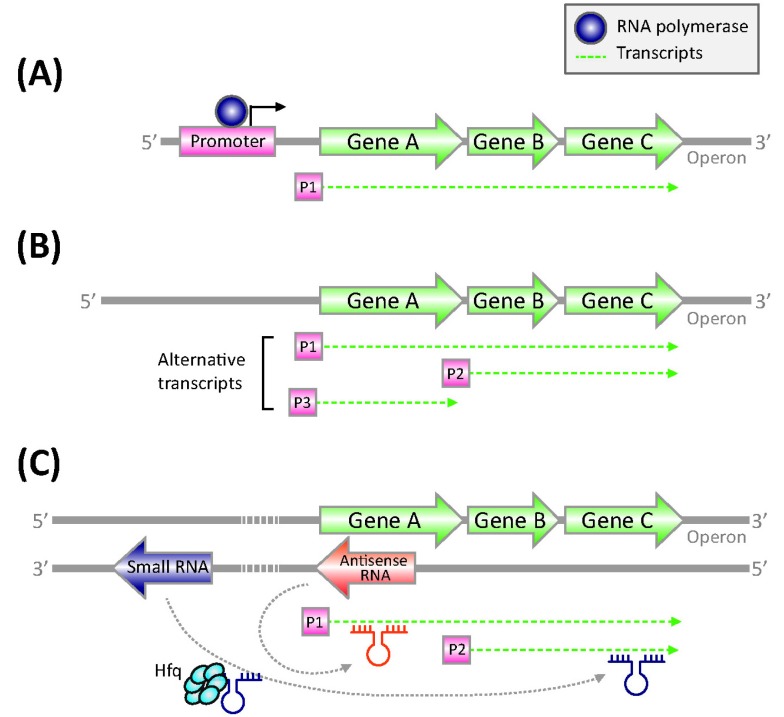
Complexity of bacterial transcriptome. (A) Operon structure of bacterial genome. (B) Definition of transcription units using alternative transcription start sites. (C) Various noncoding RNAs in regulation.
Regulatory sRNAs
In bacteria, regulatory ncRNAs have emerged as key players, acting by various mechanisms to modulate metabolic, physiological, and pathogenic processes, as well as bacterial adaptive immunity [41]. The largest group of regulatory ncRNAs consists of regulatory sRNAs, which affect the transcription, translation, and stability of target mRNAs through base pairing with the targets (Fig. 2C). These sRNAs can work in cis by overlapping their target genes encoded on the opposite DNA strand of the same genomic locus or in trans by targeting genes located elsewhere on the chromosome [41, 42]. Therefore, the discovery and characterization of bacterial sRNAs are critical for the understanding of the complexity of bacterial transcriptomes. Primary transcriptome analysis allows one to detect the presence of sRNAs at their abundance in a genome-scale by measuring nonprotein-coding RNA transcripts. For instance, an unexpected 60 sRNAs have been discovered in the primary transcriptome analysis of H. pylori, indicating that the complexity of gene expression in the small H. pylori genome is increased by genomewide antisense transcription [9]. In this way, many sRNAs were recently discovered from various bacterial species, including the opportunistic pathogen Burkholderia cenocepacia (13 sRNAs), Salmonella Typhi Ty2 (55 sRNAs), and L. monocytogenes (50 sRNAs). The sRNAs from B. cenocepacia and L. monocytogenes were induced during niche switching and involved in virulence, respectively [16, 43, 44]. Furthermore, comparing of the sRNAs with global gene expression could be facilitated in the analysis of a set of genes that are mainly involved in the pathogenic features of B. cenocepacia. In addition, immunoprecipitation of specific RNA-binding proteins can lead to selectively enriched sRNAs [8]. The first attempt at sRNA analysis, based on the NGS technique (RIP-seq), was carried out in Salmonella [29]. Using the RNA-binding property of Hfq proteins, Hfq-associated RNAs could be enriched and analyzed by immunoprecipitation of Hfq-RNA complexes, followed by deep sequencing. Through this study, more than twice as many new sRNAs were discovered over previously known sRNAs [29]. These results indicate that sRNAs are abundant in the bacterial transcriptome, representing its regulatory complexity.
Antisense RNA
RNA-seq technology has uncovered that 5% to 25% of all protein-coding genes encoded on the sense strand interact with the asRNAs that are transcribed from the antisense strand, thereby regulating their transcription, translation, or degradation (Fig. 2C) [8]. These transcripts are named asRNAs, which overlapped partially with the 5'-UTR or 3'-UTR of their target protein-coding genes [16]. The abundance of these transcripts has been revealed in several bacterial genomes, including Sallmonella Typhimurium [29], B. Subtilis [13], E. coli [45], Staphylococcus aureus [46], and M. pneumonia [6]. Using RNaseIII, which specifically degrades double-stranded short RNAs, for example, stable sRNA molecules that map symmetrically to both strands were enriched from Staphylococcus aureus. RNA-seq of the enriched RNA molecules resulted in the detection of 1387 ORFs, which covers 50% of the antisense strand [46]. Another study identified ~1,000 novel asRNAs in E. coli using high-throughput sequencing [45]. Briefly, the unique 5' end of the primary transcriptome was analyzed by treatment with tobacco acid pyrophosphatase to convert 5'PPP groups into 5'P, followed by ligation of an RNA oligonucleotide and reverse-transcription with a random primer. Further studies of several asRNAs suggested that they are involved in a wide range of cellular mechanisms, such as the translational regulation of the quorum-sensing gene rpaR in Rhodopseudomonas palustris [47]. Based on the abundance of transcripts, it has been suggested that the number of asRNAs correlates negatively with genome size [7]. Large asRNAs could be the consequence of genome reduction with transcriptional regulation, mediated at the RNA level. Taken together, RNA-seq approaches are revolutionizing our understanding of the abundance of asRNAs in bacteria [42].
Future Perspectives
With emerging advances in NGS, RNA-seq has uncovered the complexity of the bacterial transcriptome. Although transcriptional regulation by asRNAs, novel ncRNAs, and alternative splicing events have been well known in eukaryotes, similar regulatory events in prokaryotes have been newly elucidated in recent studies. In accordance with technological developments, several approaches for computational pipeline and bioinformatics analysis have been also progressed. Here, we review sequencing methods, their advantages, and several key results for bacterial transcriptome analysis. Undoubtedly, RNA-seq is having a breathtaking impact on our understanding of biological systems and will provide entirely new insights into the biological parts in living genomes. Furthermore, the concept of artificial genetic circuits has been advanced in synthetic biology, due to their simplicity in manipulating the cell [48]. For example, promoters, sRNAs, ncRNAs, and ribosome-binding sites are considered as biological parts in a cell, which can be used to assemble new genetic circuits, such as switches or oscillators. A wide range of biological parts can be screened and listed up from transcriptome analysis by NGS. Nevertheless, many efforts to develop diverse experimental approaches using NGS methods will reveal the unexpected complexity of bacterial transcriptomes.
Acknowledgments
This work was supported by the Intelligent Synthetic Biology Center of Global Frontier Project (2011-0031957, 2011-0031962), the Korea Institute of Science and Technology Information (K-13-L01-C02-S04), and the Basic Core Technology Development Program for the Oceans and the Polar Regions (2011-0021053), funded by the Ministry of Education, Science and Technology.