Introduction
Acute myeloid leukemia (AML) is one of the most wellstudied diseases in the genomic research area [1, 2]. AML occurs usually in middle-aged people and is diagnosed by increasing leukemic myeloblasts in blood over 30% [3]. AML is a genetically heterogeneous disease, since 1/3 of AML patients have chromosomal rearrangements, like t(8;21) and t(15;17), but other AML patients have normal karyotypes [4]. With recent advances of high-throughput genomic technology, a favorable prognosis has been observed with some genetic changes in cytogenetically normal AML [5]. These results were reflected by the World Health Organization (WHO) diagnostic criteria; the NMP1 and CEBPA mutations were included in the 2008 revision of these criteria [6]. The molecular change of AML is considered to be the accumulation of somatic mutations in hematopoietic progenitor cells [7]. Next-generation sequencing technology gave us new insights into the clonal heterogeneity of leukemic mutations so that we can make an explanation why some of these mutations are highly reproducible but others are very rare [8]. Still, in 30% of cytogenetically normal AML, the genetic causality origin or strongly associated genetic changes have not yet been discovered [9, 10].
With advances of high-throughput technology, discovery of disease-associated genes is growing [11]. As a consequence, the genetic knowledge databases are growing rapidly. Accordingly, the annotation of candidate causal genes in genetic studies is a very challenging process for researchers. We propose a workflow of the detection of somatic mutation candidates in 10 normal matched AML samples and introduce concept lattice analysis for clustering the samples that have highly mutated genes in common.
Methods
Primacy sequence analysis
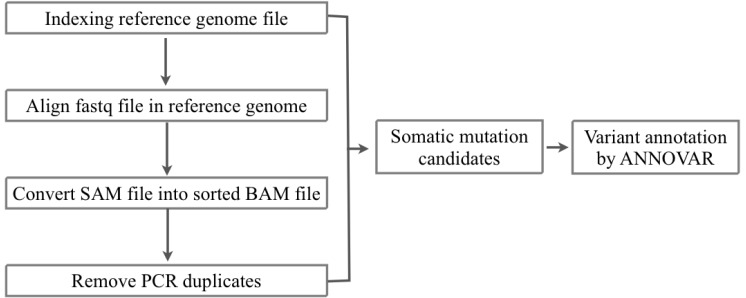
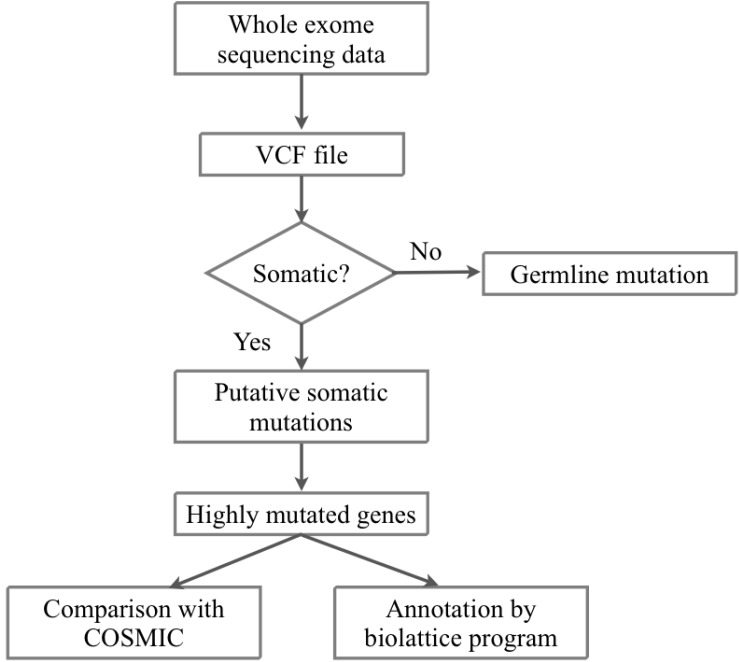
We received the fastq files of whole-exome sequencing results of tumor and matched normal sample data of 10 AML patients from the Korea Genome Organization in December 2012. There were no patient-related medical or characteristic data. We aligned the sequencing reads to the human reference genome (hg 19, GRCh37) from USCC by BWA 0.6.2 [12] (Figs. 1 and 2). To filter the known single nucleotide polymorphisms (SNPs), we used dbSNP bulid 137. We removed PCR duplicates and filtering low-quality SNPs by Samtools 0.1.18 [13], Picard 1.68 [14], and GATK 2.3.4 [15]. After the filtering process, the SAM file was converted to VCF file by VCF Tools 0.1.10 [16]. For detecting somatic mutation candidates, we obtained the difference in VCF files between tumor and normal samples. For annotation of these somatic mutation candidates, we used the ANNOVAR tool [17].
Formal concept analysis
We used formal concept analysis (FCA) for the construction of hierarchical relationships among samples sharing highly mutated genes [18]. FCA is a useful method in conceptual clustering of objects, attributes, and their binary relationship. In FCA, the sets of formal objects and formal attributes together with their relation to each other form a "formal context," which can be represented by a crosstable. In our case, the objects are 10 AML samples, and the attributes are 45 highly mutated genes. We defined the formal context as K = (G, M, I), where G is a set of objects (i.e., samples), M is a set of attributes (i.e., mutated genes), and I ŌŖå G ├Ś M is the incidence relations where (g, m) ŌŖå I if object g has attribute m. For A ŌŖå G and B ŌŖå M, we define the operators A' = {m Ōłł M|gIm for all g Ōłł A} (i.e., the set of attributes common to the objects in A) and B' = {g Ōłł G|gIm for all m Ōłł B} (i.e., the set of objects common to the attributes in B). A pair of (A, B) is a formal concept of k(G, M, I) if and only if A ŌŖå G, B ŌŖå M, A' = B, and A = B'. A is called the extent and B is the intent of the concept (A, B). The extent consists of all objects belonging to the concept while the intent contains all attributes shared by the objects. The concept of a given context is naturally ordered by the subconcept-superconcept relation, defined by (A1, B1) Ōēż (A2, B2): <=> A1 ŌŖå A2 (<=> B2 ŌŖå B1).
The ordered set of all concepts of the context (G, M, I) is denoted by C(G, M, I) and is called the concept lattice of (G, M, I). We represent the structure of this concept lattice with a Hasse diagram, in which nodes are the concepts and the edges correspond to the neighborhood relationship among the concepts. All concepts above an object label (below the attribute label) include that object (attribute). The top element of a lattice is a unit concept, representing a concept that contains all objects. The bottom element is a zero concept having no object.
Results
Overview of mutations
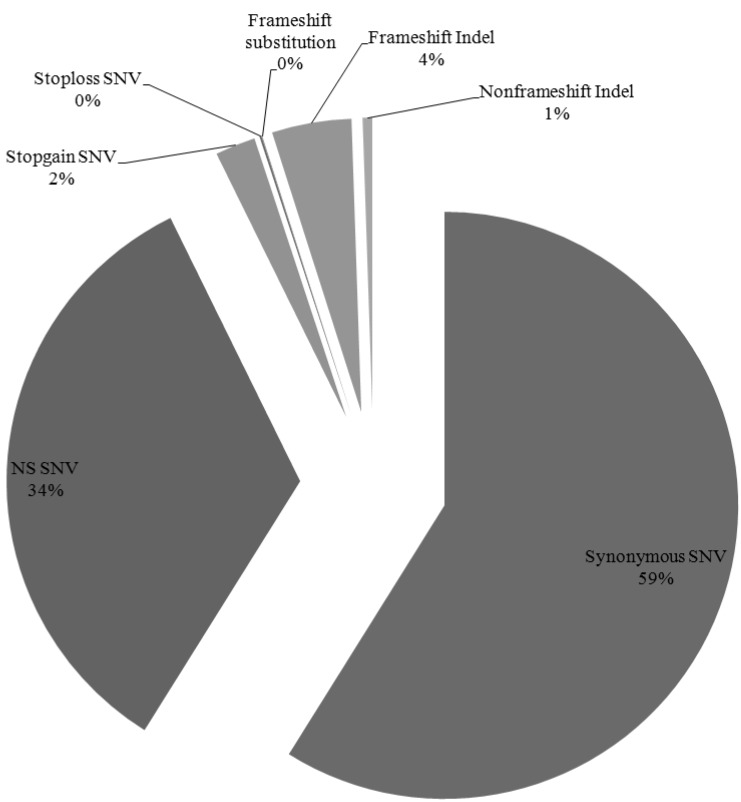
We have identified 12,908 somatic mutation candidates in 10 AML sequenced exomes, including 1,281 point mutations, 625 insertion/deletions (Indels) (Table 1, Fig. 3). The point mutations include 7,483 synonymous single nucleotide variations (SNVs), 4,297 nonsynonymous SNVs, 282 stopgain SNVs, 14 stoploss SNVs, and 5 frameshift substitutions, and the Indels include 247 frame shift insertions, 310 frameshift deletions, and 68 nonframe shifts (Fig. 4). For each patient, the average nonsynonymous mutation count was 429.7 (SD, 97.16).
About 342 to 665 genes have nonsynonymous somatic mutation candidates at least once in each AML sample (Table 2). Recurrent mutated genes were observed in all samples.
Mutation analysis
The most frequently mutated genes across all samples were USP9Y and MUC5B; these genes were mutated in 5 samples. These genes were also highly mutated in each sample; for USP9Y genes, it had 6 nonsynonymous mutations in sample 3. We have selected 45 highly mutated genes (1.5%) from 2981 mutated genes. We defined highly mutated genes as genes having 3 or more nonsynonymous mutations in each sample (Table 3). In a comparison of mutations with the COSMIC database [19], among 45 highly mutated genes, 21 genes matched to hematopoietic and lymphoid tissue malignancy terms, and 21 genes matched to other cancer types. In 3 genes, there was no matched term in COSMIC (Table 4).
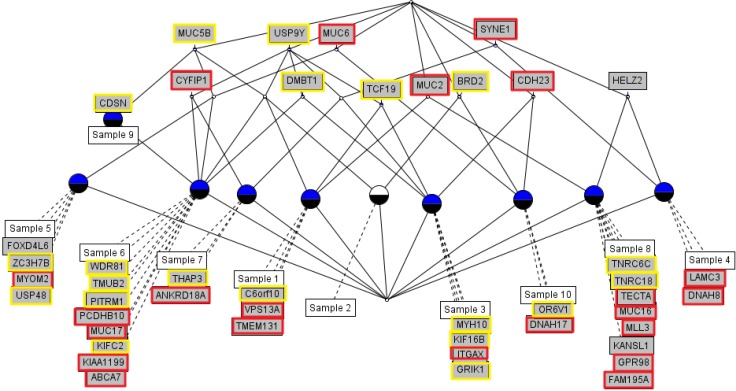
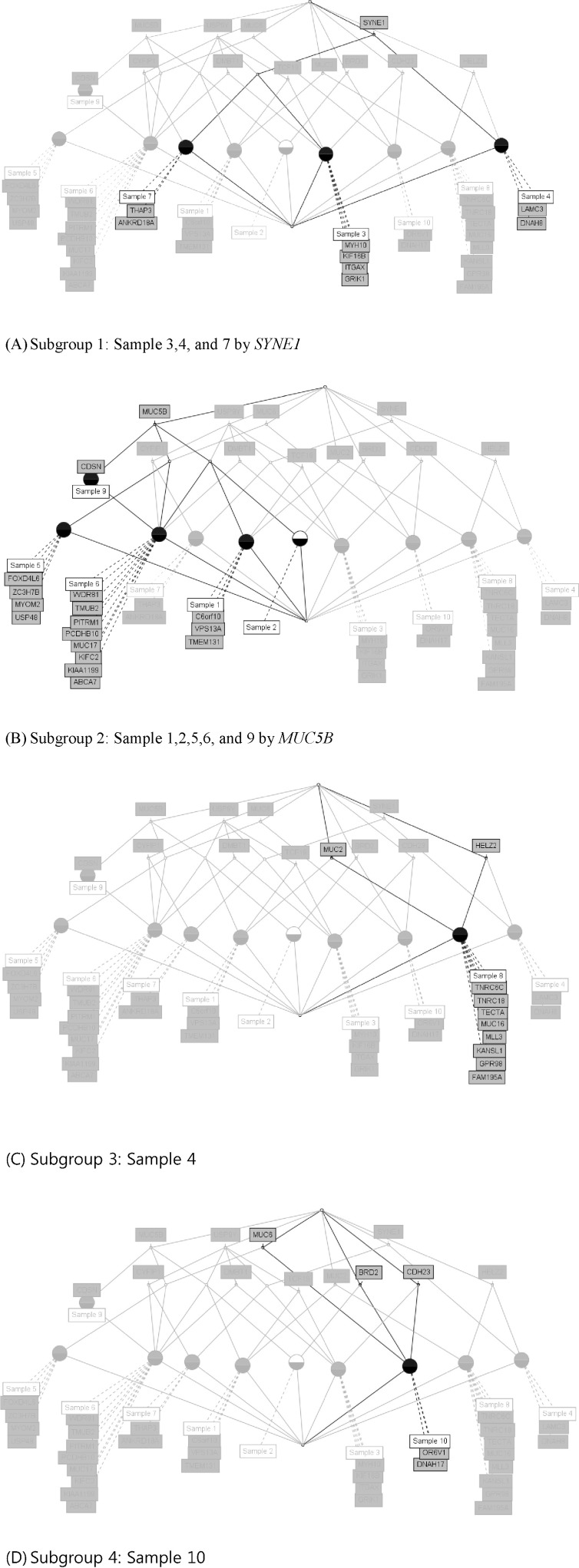
We used the concept lattice to construct the hierarchical relationship between the samples that had 45 highly mutated genes. Concept Biolattice analysis is a mathematical framework based on concept lattice analysis for better biological interpretation of genomic data. The top element of a lattice is a unit concept, representing a concept that contains all objects. The bottom element is a zero concept having no object [20, 21]. For comparing with the Concept lattice (Fig. 5), we also performed hierarchical clustering analysis by Ward method. In hierarchical clustering, cluster 1 has 5 samples (nos. 1, 2, 5, 9, and 10), cluster 2 has 2 samples (nos. 4 and 7), and others have 1 sample each (Fig. 6). We divided the samples into 4 subgroups by interpretation of the concept lattice result (Fig. 7). Lattice subgroup 1 shared SYNE1 gene mutation, and samples 3, 4, and 7 were included in this subgroup. Subgroup 2 was comprised of 5 samples (nos. 1, 2, 5, 6, and 9) that had MUC5 gene mutations in common. Samples 10 and 8 could be isolated by the uniqueness of their mutated gene sharing pattern.
Discussion
In this study, we proposed a workflow of matched normal AML exome sequencing analysis and the framework for defining sample subgroups. We observed every sample having a nonsynonymous mutation associated with hematological and lymphoid malignancy genes, but the candidate oncogenes showed diverse characters.
We selected 45 genes that had 3 or more nonsynonymous mutations and performed hierarchical clustering analysis of the samples by these genes. In classic hierarchical clustering analysis by Ward's method, we could not identify the genetic relationship of those clusters. On the other hand, the result of concept lattice analysis gave us insight into the mutational pattern of each sample.
In subgroup 1, samples 3, 4, and 7 shared SYNE1 gene mutations. SYNE1 gene encodes a spectrin repeat-containing protein expressed in skeletal and smooth muscle and peripheral blood lymphocytes that localizes to the nuclear membrane [21]. This gene is not a well-known leukemic gene but is observed in some hematological malignancies and other cancer types [22]. In glioblastoma, SYNE1 mutation is significantly correlated with the overexpression of several known glioblastoma survival genes [23]. In the case of sample 3, the ITGAX gene, encoding ankyrin repeat domain 18A, was mutated. This gene is well known by the association with leukemia [24] and lung cancer [25]. For sample 4, the possible oncogene is LAMC3. LAMC3 gene encodes laminins, which are the major non-collagenous constituent of basement membrane. LAMC3 mutations are associated with several cancers, including colon cancer, lung cancer, and melanoma, and candidate tumor suppressor genes in bladder transitional cell carcinoma [26]. LAMC3 is involved in the phosphatidylinositol 3-kinase.Akt signaling pathway, since it has a role in cell adhesion. The ANKRD18A gene is the oncogene candidate for sample 7 and is a novel epigenetic regulation gene in lung cancer [25]. Therefore, it is possible that the pair relationship of those genes (ITGAX-SYNE1, LAMC3- SYNE1, and ANKRD18A-SYNE1) could contribute together to evolve the leukemic cell transformation.
The major limitation of our study is that we could not validate the mutation results by Sanger method or deep sequencing. We selected highly mutated genes having 3 mutations or more, but this definition is arbitrary, so we might have lost candidate oncogenes in some patients.
In this study, we suggest the concept of clustering samples that have diverse mutated genes. AML is very heterogeneous genetic disease. Despite the small number of samples we have studied, the genetic variation patterns were not common for all samples. It could have been better to evaluate more sample data for analysis by clustering analysis.