Introduction
Hearing impairment is the most common neurosensory disorder [1] in humans. As of global statistics, approximately 360 million people have been suffered from hearing loss (HL) [2]. As a result, HL is reported as the fourth leading cause of disability in humans worldwide [3]. Meanwhile, congenital HL appears as the most prevalent chronic condition and defect in children [4,5]. The growing cases of hearing impairment in newborn are also reported by the universal newborn hearing screening board [5]. It is also identified that 70% of congenital HL accounts for non-syndromic hearing loss (NSHL) [6] whereas the rest 30% comes under syndromic [7]. As per literature, molecular and genetic aetiology is the prime cause of congenital HL. But, due to genetic and clinical heterogeneity in NSHL, the disease pathogenesis [6] is not clearly understood. Therefore, there is a subsequent delay in the discovery of disease pathways and identification of molecular targets since 1990 [8]. Again, complexity in the identification of potential target genes and their associated variants at an accurate genomic location has created lots of diagnostic complications in NSHL [9].
The association of several important genes such as GJB2, GJB3, GJB6, SLC26A4, KCNQ4, DFNA5, SLC26A5, MYO1A, MYO7A, MYH15A, and CDH23 has been identified with respect to the progression of congenital NSHL. Among these, GJB2 is established as an imperative target of NSHL [9]. In addition, the successful adoption of genome-wide association studies in biomedical research has discovered the genetic architecture of novel variants and their disease associations [10,11] which encourages researchers to focus on the identification of a potential biomarker for NSHL.
Generally, identification of potential biomarkers through the integration of multiple complementary approaches [12] is better than using any single method. As of evidence, many studies have been reported using more than one in silico methods such as pathway enrichment and functional annotation or protein interaction (PPI) network to screen and/or discover potential target genes from a large scale gene pool in diseases like atherosclerosis, nasopharyngeal carcinoma, systemic sclerosis, Parkinson disease, papillary thyroid carcinoma, neurodegeneration, etc. [13-19]. So, to identify any putative target genes, different aspects including the study of different regulatory pathways, miRNA-based gene regulation, annotation of protein functional network, and variant analysis are need to be addressed. Particularly, the discovery of important biological pathways involved in signal transductions [20] has a great aid and value. In this connection, the study of functionally important protein-PPI provides a meaningful direction towards understanding the involvement of disease targets in different biological processes [20]. Similarly, the discovery of miRNAs-based gene regulatory networks in the post-transcriptional process has a certain importance in biomarker identification [21]. As proof, experimental studies have also shown the involvement of a few miRNAs (miR-182, miR-183, and miR-96) in human inner ear development [22-25]. In addition to this, detection of deleterious single nucleotide polymorphisms (SNPs) has also been implemented in biomarker identification to ascertain disease susceptible genes. Especially, non-synonymous SNPs in the coding region have a vital role to produce various damaging effects on protein structure, stability, charge, etc., which leads to functional dysfunction of several disease targets [26-28]. Therefore, the identification of non-synonymous SNPs within the proper coding architecture of disease targets is an important strategy of biomarker identification. The present study is focused on the discovery of potential NSHL targets and their functional variants using multiple silico complementary approaches.
Methods
Data mining
A systematic review of literature was carried out by following the guidelines of Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA). The scientific articles on NSHL were searched in PubMed (https://pubmed.ncbi.nlm.nih.gov/), Science Direct (https://www.sciencedirect.com/), Cochrane Library (www.cochranelibrary.com/), and JSTOR (www.jstor.org/) databases using MeSH terminologies such as âNon syndromic hearing lossâ (congenital non syndromic hearing loss* (MeSH), non-syndromic hearing loss* (MeSH), non-syndromic hearing loss (MeSH)) and âgeneticsâ (gene* (MeSH), genetics* (MeSH)), AND âepidemiology & pathogenesisâ (epidemiology* (MeSH), pathogenesis*(MeSH)). The articles were collected from the last decade up to December 2020. Following inclusion and exclusion criteria were followed to select the appropriate literature for reference.
Inclusion criteria
The following criteria were followed for selecting the articles:
(1) Literatures providing information about target genes involved in NSHL.
(2) The articles published in the English language.
(3) The full-length article available from period 2009-2020.
All available full-length original research articles and case reports were included.
Exclusion criteria
The rest of the literatures were excluded based on the following exclusion criteria:
(1) The review articles, abstracts, articles written in other languages, letters to the editor, short reports, and correspondences.
(2) The articles are based on syndromic HL and the adult population.
(3) Articles without any genetic information.
Dataset preparation
The NSHL target genes were selected from shortlisted literatures and subjected for removal of duplicate genes. The unique NSHL target gene list was prepared for further analysis.
Discovery of NSHL pathway
All of the selected genes were employed to discover their association in different biochemical and signaling pathways involved in the development and progression of NSHL using KEGG (Kyoto Encyclopedia of Genes and Genomes) Mapper web server (https://www.genome.jp/kegg/mapper.html).
Functional network analysis
The pathway associated NSHL target proteins were analyzed using STRING (https://string-db.org/) web server in order to represent a functional network between them.
Discovery of regulatory miRNAs-gene interaction in NSHL progression
Functionally associated target genes were subjected to miRTarBase (http://miRTarBase.cuhk.edu.cn/) web server to analyze their regulatory interaction with three previously reported miRNAs (miR-182, miR-183, and miR-96). The identified miRNA-gene interaction hubs were validated and graphically represented through miRNet (https://www.mirnet.ca/) tool.
Variant analysis
Effective SNPs were predicted from NSHL target genes to identify their functional variants. SNPs were searched from dbSNP database (https://www.ncbi.nlm.nih.gov/snp/). Validation of SNPs were performed using SIFT (https://sift.bii.a-star.edu.sg/) [29], PredictSNP1 (https://loschmidt.chemi.muni.cz//predictsnp1/), and PredictSNP2 (https://loschmidt.chemi.muni.cz/predictsnp2/) algorithms [30].
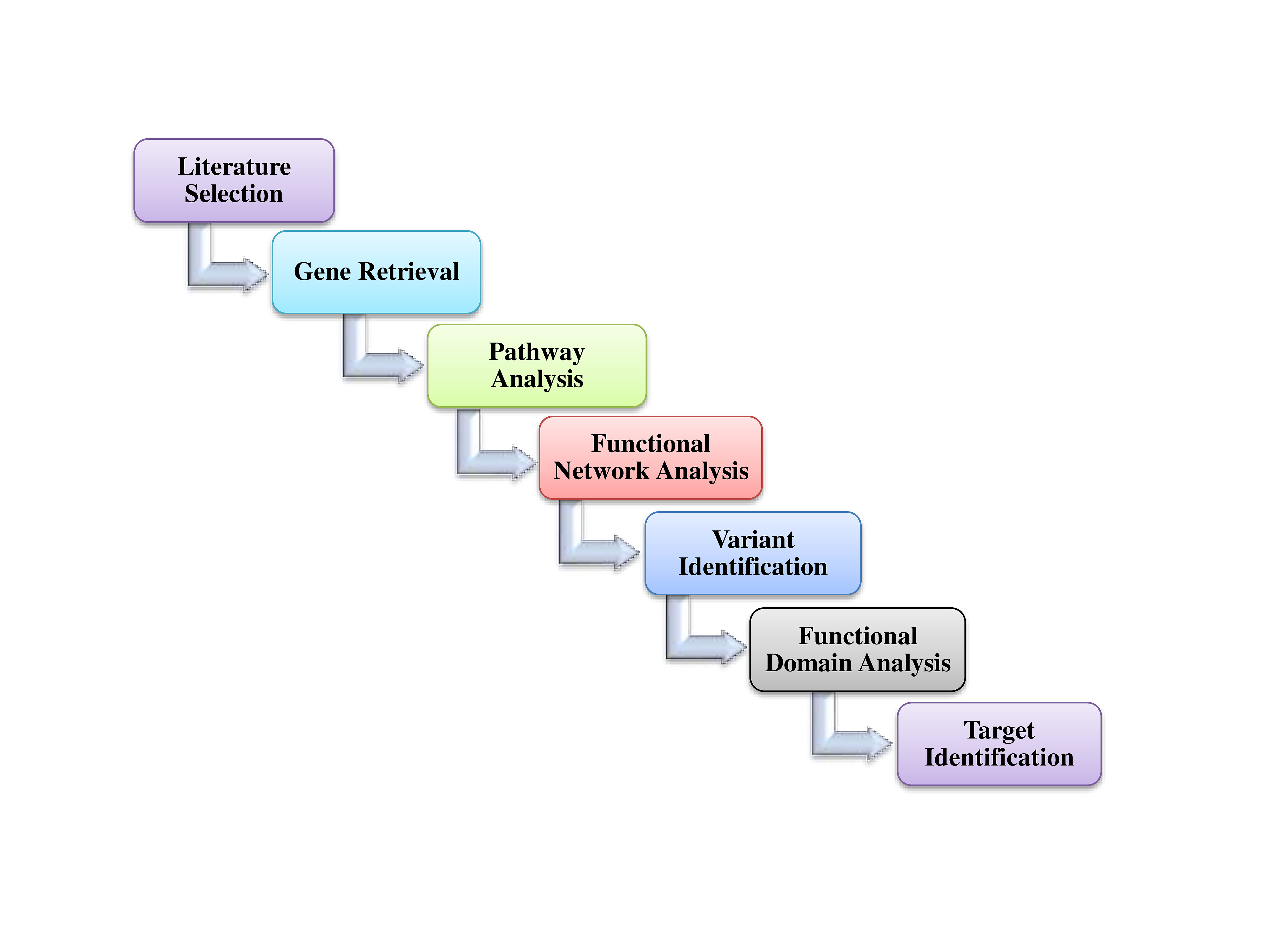
The schematic representation of overall procedures is depicted in Fig. 1.
Results
Selection of target genes
Total 14,215 numbers of articles were selected on the basis of PRISMA guidelines from different scientific repositories such as PubMed (3,172), JSTOR (209), Science Direct (10,827), and Cochrane (7). Among these, a total of 787 studies were satisfied with all of the inclusion criteria and considered for this study (Fig. 2), whereas the remaining studies were excluded as per the exclusion criteria manually. Further, a total of 2,707 NSHL target genes were collected from these 787 studies, out of which 422 genes were identified as unique. Again, from these 422 unique target genes, mitochondrial RNA (5), DNA (1), and microRNAs (7) were discarded, and the rest of 381 genes (Fig. 2) were considered as the final data set of NSHL targets.
Analysis of NSHL target genes
The association of 381 selected genes was searched against different biological pathways to discover potential NSHL targets. It was interpreted, a total of 23 genes (Fig. 2) are significantly involved in six different pathways such as Notch signaling pathway (hsa04330), Wnt signaling pathway (hsa04310), gap junction (hsa04540), tight junction (hsa04530), JAK-STAT signaling pathway (hsa04630), and adherens junction (hsa04520) which are associated with NSHL development (Table 1). Therefore, these 23 genes such as NOTCH1, RBPJ, LRP5, SMAD4, RAF1, ADCY1, GJA1, SNAI2, ACTB, ACTG1, FGFR1, MET, TJP2, CLDN14, CLDN9, MARVELD2, MYH9, NEDD4, NF2, RDX, IFNLR1, IL13, and PTPN11 were anticipated as potential NSHL targets. Afterward, functional association of these 23 NSHL targets was studied using the STRING tool with the high level of confidence parameter (score 0.007). The resulting protein-protein network showed a strong interaction between 16 nodes (genes) (Fig. 3) with a significant PPI enrichment p-value of 0.00107. Particularly, strong functional interactions have been observed between six groups of targets i.e., RBPJ, NOTCH1, SNAI2, and SMAD4; RDX, ACTB, ACTG1, and MYH9; CLDN9-CLDN14; TJP2-MARVELD2; PTPN11-MET; NEDD4-GJA1 in the resulted PPI network. Further, the involvement of these 16 NSHL target genes was also confirmed in a few hearing associated biological processes such as inner ear development, inner ear auditory receptor cell differentiation, homeostatic process, chemical homeostasis, signal transduction, regulation of response to external stimulus, related to hearing development and impairment (Table 2).
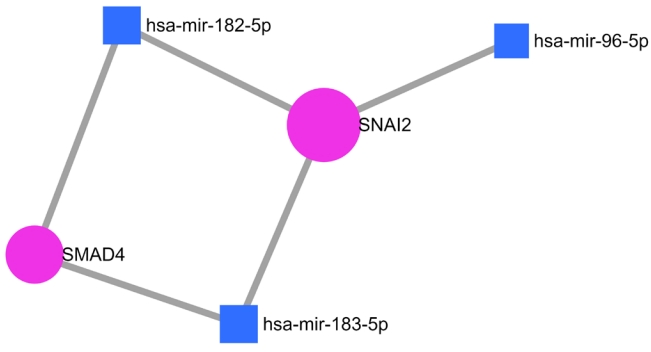
Further study was performed to analyze the gene regulatory network between these 16 NSHL target genes with three important miRNAs such as miR-182, miR-183, and miR-96 which are significantly expressed in the human inner ear and associated with NSHL. Among these, the strongly validated interaction was found between the target genes SMAD4 and SNAI2 with miR-182 and miR-183, whereas miR-96 regulates only SNAI2 (Fig. 4).
Identification of deleterious variants in NSHL target genes
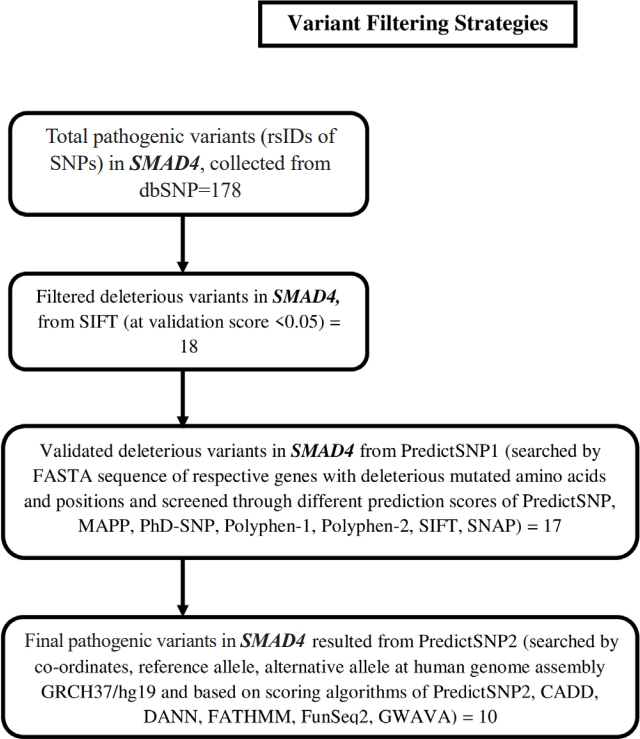
Non-synonymous SNPs (nsSNPs) were predicted for two important NSHL targets (SMAD4, SNAI2) confirmed from previous analysis. The prediction was resulted total 178 rsIDs of pathogenic variants for SMAD4 gene with clinical significance from dbSNP (Supplementary Fig. 1). At the same time no pathogenic variant was obtained for SNAI2 gene. Initial validation using SIFT algorithm (<0.05) was identified 18 rsIDs (out of 178 rsIDs) in SMAD4 with deleterious effect (Supplementary Fig. 2). Afterwards, cross validation using PredictSNP1 was resulted 17 rsIDs in SMAD4 (Supplementary Fig. 3) as deleterious variants. Finally, theses variants were further validated using PredictSNP2 algorithms which was resulted 13 numbers of deleterious nsSNPs (Table 3, Fig. 5) assigned with 10 rsIDs i.e., rs377767345 (G352E), rs121912581 (G352R), rs377767347 (R361H, R361L), rs377767348 (C363R), rs121912580 (G386D), rs377767367 (G395V, G491V), rs377767369 (W509R), rs377767371 (G510V), rs377767382 (L533P, L533R), rs377767381 (L533V).
Furthermore, functional domain regions of SMAD4 were analyzed from the respective protein structure. Interestingly, all of these 13 deleterious variants (Table 3) were found to occur within its functional domain i.e., MH2 domain (323â552 AA) of SMAD4 gene (Fig. 6). Therefore, the present observation has added remarkable evidence to this study for putative NSHL target identification.
Discussion
Heterogeneity in NSHL is still under the puzzled cube which has encouraged researchers to understand its accurate genetics. In the present study, authors have attempted to discover potential NSHL target genes and their functional variants using a computational approach. Initial prediction has identified the involvement of 23 genes in the inner ear development and hearing impairment pathways such as Notch signaling pathway, Wnt signaling pathway, gap junction pathway, adherens junction pathway, tight junction pathway, and JAK-STAT pathway. As per the literature evidence, all of these six pathways are appeared to have a crucial role in the NSHL progression. Several studies have been reported about the differential expression pattern of different functional genes in Notch signaling pathway which has multiple roles in the development of inner ear including regulation of hair cell, determination of neurons, sensory regions, and non-sensory regions [31]. It is also disclosed that, Wnt signaling pathway has a dominant role in the dorsal structure formation of the inner ear [32,33]. In addition, a critical role in hearing is directed through gap junction pathway and mutations in connected genes have been reported to cause a high incidence of human deafness [34]. Similarly, several physiological processes such as cochlear development, growth of auditory neurons, immune mediation, and planar cell alignment are maintained through Adherens junction pathway [35]. Again, in the inner ear, tight junction is important for the separation of endolymphatic and perilymphatic space to maintain the concentration gradient between endo and perilymph and the endocochlear potential [35,36]. Mutations in genes and/or proteins associated with this pathway are reported to cause hereditary HL [35,37]. Likewise, JAK-STAT signaling pathway balances the noise-induced damage to cochlear tissue and loss of hearing sensitivity. The imbalanced expression of STAT3 is caused by loud sound which leads to cochlear tissue damage and loss of hearing sensitivity [38]. All of this evidence stood in support of 23 genes as potential NSHL targets identified from the present study.
Afterward, participation of 16 genes (out of 23) in different biological processes such as inner ear development, homeostatic process, chemical homeostasis, signal transduction, inner ear auditory receptor cell differentiation, and regulation of response to external stimulus related to hearing impairment and NSHL progression was elucidated through study of their PPI network [39-41]. Therefore, all of these biological processes are providing the major key points to consider the involved genes as potential NSHL targets. The predicted strong functional association between these genes also supported the above hypothesis. From subsequent analysis, a strong regulatory interaction between two important NSHL target genes (SMAD4 and SNAI2) with three established miRNAs (miR-183, miR-182, and miR-96) was obtained which have added a worthy value to this current study. The expressions of these three miRNAs in the human inner ear and their involvement in NSHL progression [15-18] have been studied well. According to the previous findings, both of these miRNAs (miR-182 and miR-183) down-regulate the SMAD4 expression in human bladder, prostate cancer, and ovarian cancer [42-44]. Apart from these several others such as miR-183 regulates ATP2B4, BTG1, EZRIN, GNG5, KCNJ14, NCS1, NEFL, PEX19, PPP2CA, SLC6A6, TREK-1, ZCCHC3 [18] and miR-182 regulates CaV1.2, MITF, RDX [15,18]. Similarly, miR-96 is known to regulate several genes including MYRIP, AQP5, CaBP1, CaV1, CELSR1, CELSR2, GRID1, KCC2, MITF, RDX, RYK, and TFCP2L3 [17,18]. At the same time, a study in the mice model has confirmed the regulation of SNAI2 gene by miR-96 [45].
As, the regulation of SMAD4 and SNAI2 through miR-183, miR-182, and miR-96 in human NSHL is still not clear, further investigation may provide proper direction into it.
Here, total of 13 deleterious nsSNPs (G352E, G352R, R361H, R361L, C363R, G386D, G395V, G491V, W509R, G510V, L533P, L533R, and L533V) were obtained within the functional region i.e., MH2 domain of the SMAD4 gene. SMAD4 is a multifunctional modulator, which regulates cell proliferation, differentiation, autoimmunity, pluripotency, plasticity, cell growth, apoptosis, autophagy, invasion, and metastasis. Deregulation of SMAD4 is also associated with embryonic developmental diseases [46]. It has a vital role in the activation of Wnt signaling pathway [47]. The SMAD4 variants G352E [48], G352R [49], R361H, R361L [48], C363R [50], G386D [49], W509R, G510V [50], L533P, L533R [48], and L533V [51] are previously identified in juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome, whereas G491V is reported in colorectal cancer [51]. The appearance of most of these deleterious mutations in the MH2 domain of SMAD4 protein has emphasized their importance for further study. MH2 domain is a multifunctional region that mediates differential interaction with other proteins, transcription factors, co-activators, and co-repressors. These interactions provide specificity and selectivity to SMAD4 functions [52,53].
Interestingly, the association of a deleterious variant i.e., rs377767367 (G491V) in NSHL target SMAD4 was discovered and is not reported in the NCBI ClinVar database for any disease conditions. Thus, present findings will not only provide insights into the genetics of NSHL but also help in further genetic counselling of NSHL after subsequent investigation.
NSHL is a critical hereditary disorder. Complexity in disease diagnosis increases due to its clinical and genetic heterogeneity. The present study has reported SMAD4 as the most potential target gene in the HL development pathways. The discovery of important miRNAs, miR-183, and miR-182 in associated with the regulation of SMAD4 has also supported the above hypothesis. Identification of pathogenic and deleterious nsSNPs within its functional domain is also added an additional clue to this study. Outcomes of the present investigation may be useful in understanding the genetics of NSHL and also throw light on the diagnosis modalities for NSHL. All of these findings are needed to experiment with for further validation.