Introduction
â Acquisition: Acquiring SARS-CoV-2 RNA sequences
â Preparing: It consists of exploring and pre-processing data in such a way that only valid and adequate data are kept for appropriate analysis.
â Analyze: This is the step where we are leveraging on acquired skills in HPC, and distributed computing in general, to optimize the processing. The analysis in this work is divided into three steps that we are detailing later in this paper.
â Report: Once the analysis is done, the remaining challenging problem would be to visually present the results in an efficient and readable way to explain to the action makers what should be done to improve the results in terms of biological value.
â Action: After presenting the blueprint for an open-source HPC and LCS-based platform for SARS RNA clustering. This can help researchers in medicine and virologist in clustering and identifying the rapid variance in SARS-CoV-2.
Main Text
Sequence alignment
Problem statement
HPC infrastructure
Hardware
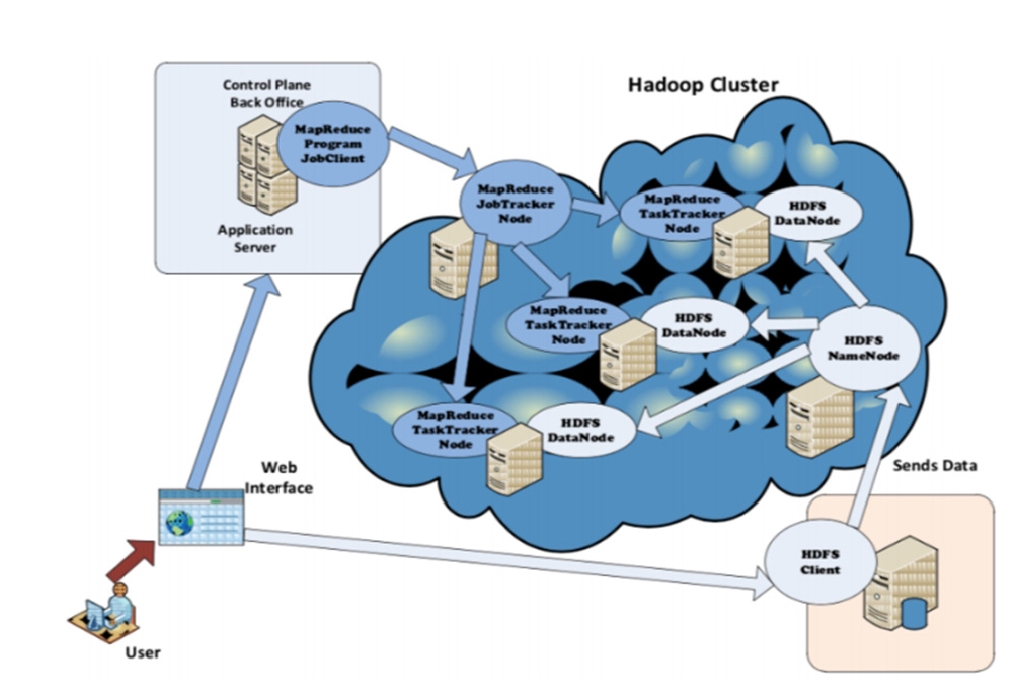
Hadoop deployment
â Namenode, which is the master node whose task is to manage the distributed file system by keeping relevant metadata and namespace entries.
â Datanodes (workers), which store and extract blocks upon requests from the Namenode.
â JobTracker: Coordinates job execution by splitting the main job into tasks, and delegates them to the TaskTrackers while considering two essential factors: load balancing and location of the chunks in the HDFS Datanodes.
â TaskTracker: The MapReduce horse-workers that run the tasks assigned by the JobTracker.
â HDFS: Responsible for providing the chunks to the TaskTrackers.
Proposed approach
DS step 1: data retrieving
DS step 2: data preparing
DS step 3: analysis
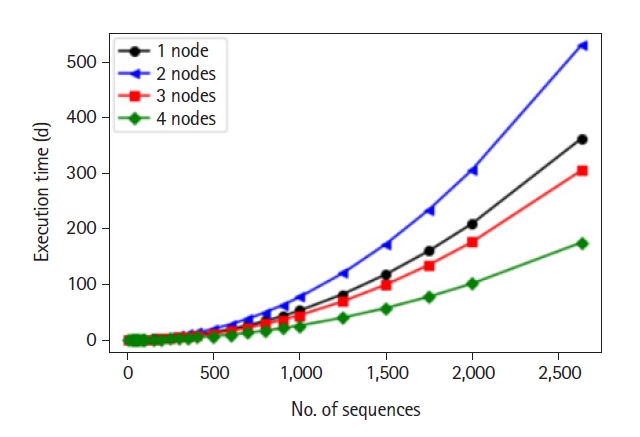
Length of LCS computing
â With 30 RNAs, we performed (30 * 29)/2 = 435 comparisons and it took about 59 min to finish.
â With 2.635 samples, we would need about 1 y and this is where HPC comes into play and proves indispensable in bioinformatics.