Rapid and sensitive detection of Salmonella species targeting the hilA gene using a loop-mediated isothermal amplification assay
Article information

Abstract
Salmonella species are among the major pathogens that cause foodborne illness outbreaks. In this study, we aimed to develop a loop-mediated isothermal amplification (LAMP) assay for the rapid and sensitive detection of Salmonella species. We designed LAMP primers targeting the hilA gene as a universal marker of Salmonella species. A total of seven Salmonella species strains and 11 non-Salmonella pathogen strains from eight different genera were used in this study. All Salmonella strains showed positive amplification signals with the Salmonella LAMP assay; however, there was no non-specific amplification signal for the non-Salmonella strains. The detection limit was 100 femtograms (20 copies per reaction), which was ~1,000 times more sensitive than the detection limits of the conventional polymerase chain reaction (PCR) assay (100 pg). The reaction time for a positive amplification signal was less than 20 minutes, which was less than one-third the time taken while using conventional PCR. In conclusion, our Salmonella LAMP assay accurately detected Salmonella species with a higher degree of sensitivity and greater rapidity than the conventional PCR assay, and it may be suitable for point-of-care testing in the field.
Introduction
Salmonella species are among the major pathogens that cause foodborne infections. Indeed, Salmonella species are thought to be the main factor contributing to diarrheal disease-associated deaths [1]. According to the World Health Organization, Salmonella accounts for approximately 9% of diarrheal illnesses and it costs 2.7 billion US dollars to care for these patients in the United States per year [2,3]. Especially, since food production is evolving to reflect demand for raw and lightly cooked foods, foodborne pathogens including Salmonella have continued to spread all over the world [4,5].
To detect Salmonella at the early stage of infection, a sensitive and reliable method is crucial. Various methods have been developed for Salmonella detection using molecular tools such as direct polymerase chain reaction (PCR) and multiplex quantitative PCR [6]. A PCR-based assay is currently the most commonly used tool for molecular screening of Salmonella [7]. However, PCR methods require specialized equipment that must be optimized for the amplification of target genes from samples, along with the extraction and purification steps carried out by a well-trained individual. Several technical issues such as long reaction times (about 2 h) and the requirement for well-purified nucleic acid are drawbacks for clinical application in many real-world settings. The lack of hardware resources in the field constitutes a critical barrier to the detection of pathogens directly from clinical samples [8]. Taken together, the limitations of conventional PCR-based methods make these assays difficult to use for point-of-care testing (POCT). POCT is a new concept in the laboratory-medicine discipline that enables screening for pathogens at the site where the infection occurs without transporting the test materials to well-equipped laboratories; by obviating the need for transport, POCT would be helpful to reduce the time necessary for clinical decision-making [9,10]. Therefore, POCT requires fast reaction time and simple equipment.
Loop-mediated isothermal amplification (LAMP) technology, an isothermal nucleic acid amplification method developed by Notomi et al. [11], can amplify target nucleic acids at a consistent temperature without changing the temperature for PCR cycling. The LAMP assay has been proven to be an effective tool for POCT in the field to test for infectious diseases [12-14]. There are several advantages of the LAMP technology. First, due to isothermal amplification, LAMP does not need a thermal cycler; therefore, this technology is ideal for POCT in the field. Second, the reaction time is much shorter than that of PCR, which is advantageous for rapid testing of highly contagious infections and preventing the spread of infections at an early stage. Third, the detection sensitivity of LAMP is higher than that of conventional PCR due to the loop-mediated amplification strategy. Fourth, LAMP is relatively less sensitive to PCR inhibitors, which can contaminate samples from sources such as feces; therefore, LAMP would be advantageous for minimizing the sample preparation procedure. During the LAMP reaction, the inner primers anneal by Watson-Crick complementarity to a region within the target and a hybridized loop structure is generated by strand invasion, which allows more efficient amplification for the synthesis of large amounts of DNA in a short time [11].
Several studies have reported LAMP assays targeting Salmonella in food [15,16]. The invA gene has been the most frequent target for Salmonella detection LAMP assays [15]. In this study, we aimed to develop a real-time LAMP assay targeting the hilA gene for universal detection of Salmonella species. We also validated the LAMP assay with seven clinically important Salmonella pathogens.
Methods
Bacterial strains
In this study, seven major Salmonella species were used. Six species (Salmonella Typhi, Salmonella Typhimurium, Salmonella enteritidis, Salmonella Paratyphi A, Salmonella Paratyphi B, and Salmonella Infantis) were collected from the Korea National Institute of Health (KNIH), Republic of Korea. Salmonella enterica was collected from Chungbuk Veterinary Service Laboratory, Chungju, Republic of Korea. For the specificity test, we used 11 non-Salmonella pathogens: seven Gram-negative and four Gram-positive bacteria (Table 1).

Strains used for testing the Salmonella LAMP assay based on the hilA gene
DNA extraction and template DNA preparation
All Salmonella strains (n=7) were cultivated for 24 h at 37℃ on sheep blood agar (Bandio, Pocheon, Korea). Non-Salmonella strains (n=11) were cultivated for 36 hours at 37℃ on Difco Luria-Bertani agar (BD, Franklin Lakes, NJ, USA). DNA extraction was done via QlAamp DNA Mini kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions using the protocol. For Gram-negative bacteria, colonies on the freshly cultured bacteria were added to the ATL buffer (180 μL) containing proteinase K (20 μL; 20 mg/mL) and suspension. The sample was then mixed by vortexing and incubated at 56℃ until the bacteria were completely lysed. Next, 200 μL of lysis buffer was added to each sample and incubated at 70℃ for 10 min. Then, vortexing for 15 seconds was performed right after adding 200 μL of ethanol. The mixture was transferred to the QlAamp Mini Spin column and centrifuge at 6,000 ×g for 1 min. The genomic DNA in the spin column was washed using 500 μL of AW1 buffer and centrifuged at 6,000 ×g for 1 min. Next, 500 μL of AW2 buffer was added and centrifuged at 20,000 ×g for 3 min. For the elution process, 100 μL of distilled water was added and centrifuged at 6,000 ×g. The genomic DNA was measured using Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and the DNA was stored at ‒20℃ for further study. Gram-positive bacteria were added to 180 μL of lysis buffer containing proteinase K (20 μL; 20 mg/mL) and incubated for 30 min at 37℃ instead of using the ATL buffer. The other steps were the same as in the protocol for Gram-negative bacteria.
Primer design for the Salmonella LAMP assay
Primer Explorer V4 (http://primerexplorer.jp/elamp4.0.0/index.html) was used to design Salmonella species LAMP primers. The three sets of primers included a forward outer primer (F3), a backward outer primer (B3), a forward inner primer (FIP), a backward inner primer (BIP), and two loop primers: a forward loop primer (LF) and a backward loop primer (LB). All sets of primers were then validated by the BLAST program. Of the three sets of LAMP primers, the set that demonstrated the best amplification performance was selected as the Salmonella LAMP primer set (Fig. 1).
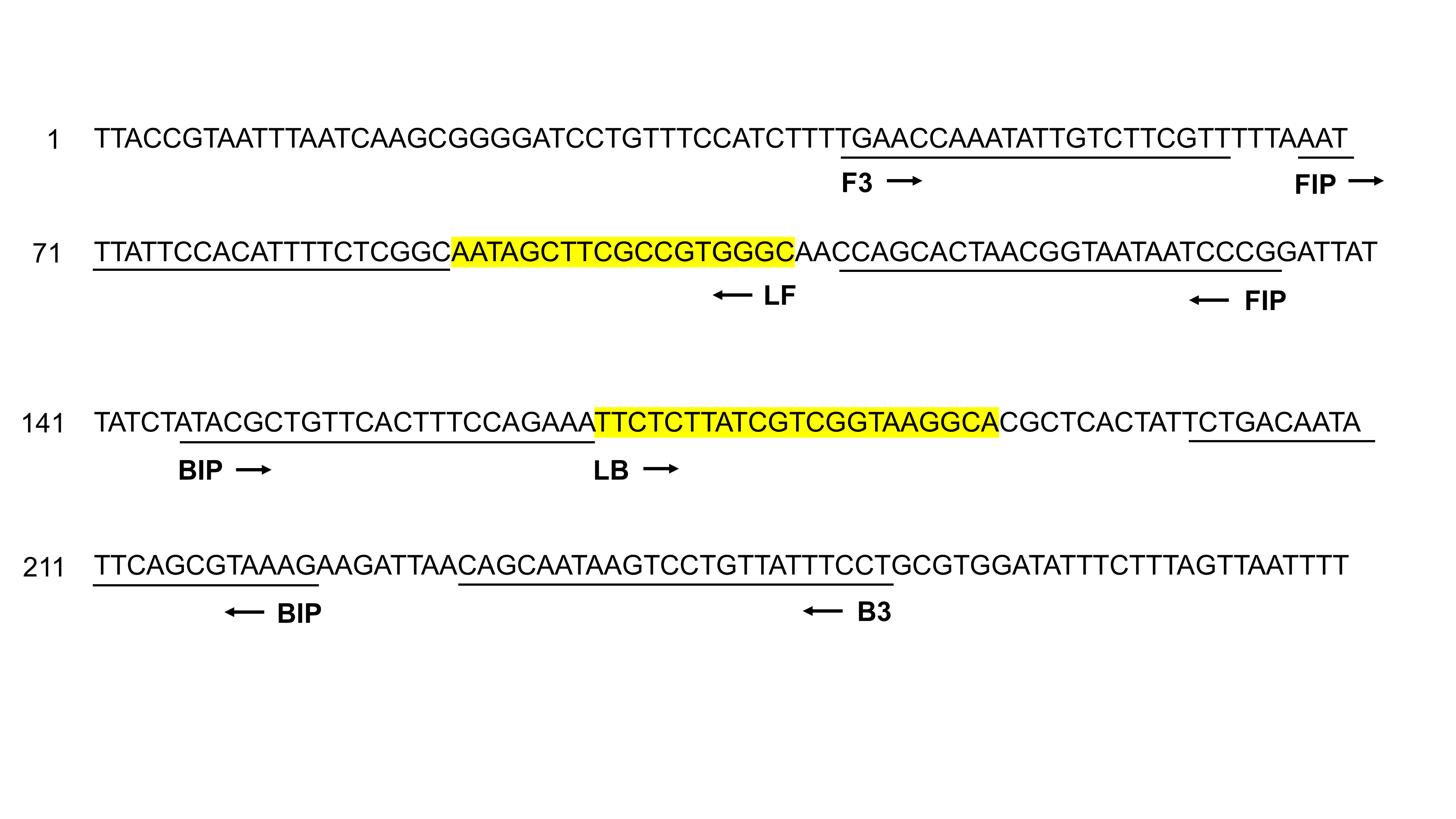
Primer design for the Salmonella loop-mediated isothermal amplification assay. The positions of six primers (F3, B3, FIP, BIP, LB, and LF) were aligned on the partial nucleotide sequence of the hilA gene (GeneBank accession No. CP075108). The forward/backward outer primers (F3/B3) and forward/backward inner primers (FIP/BIP) are underlined. The forward/backward loop primers (LF/LB) are highlighted in yellow. Arrowheads indicate the direction of the primers.
LAMP assay
The LAMP reaction was carried as described elsewhere with some modifications [17]. In brief, a 20 μL reaction mixture was prepared that contained 1 μL of target genomic DNA along with 1.6 μM each of the primers FIP and BIP, 0.2 μM each of F3 and B3, 0.4 μM of LF and LB. 4U of the Bst 2.0 DNA polymerase (New England Biolabs, Ipswich, MA, USA), 8 mM of MgSO4 (New England Biolabs), 1.5 mM of dNTPs (Thermo Fisher Scientific), 1× isothermal amplification buffer (New England Biolabs), and 1.25 M of N-methyl formamide (NMF) and isobutylamide (IBA). Furthermore, 2 μM of SYTO 9 (Thermo Fisher Scientific) was added to enhance fluorescence in the presence of DNA in the real-time assay. The amplification reaction was performed at 60℃ for 45 min and terminated by heating at 80℃ for 3 min using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). The results were shown as a graph on the monitor of real-time analysis software (BioRad CFX Manager, Bio-Rad Laboratories). The fluorescence curve was captured from the BioRad CFX Manager graph.
Optimization of the LAMP reaction and evaluation of the detection sensitivity and specificity
To optimize the amplification conditions, the LAMP reaction was performed at different temperatures (60℃–65℃). To evaluate the detection sensitivity of the LAMP assays, we tested the detection limit using S. enterica DNA, which was 10-fold serially diluted from 1 ng to 1 ag and used in the LAMP reaction. To test for non-specific amplification of non-Salmonella pathogens, we conducted the LAMP assay with seven non-Salmonella Gram-negative and four Gram-positive bacteria.
Results
Design of LAMP primers
Six LAMP primers were designed targeting the hilA gene as a universal marker of Salmonella species: two outer primers (F3 and B3), two inner primers (FIP and BIP), and two loop primers (LF and LB) (Fig. 1). Validation of the primer set was performed using S. enterica DNA under standard LAMP conditions (60℃ for 45 min) (Fig. 2). We performed the LAMP reaction with four different combinations of primers. Set 1 involved LAMP with two inner and two outer primers but without a loop primer, set 2 used the set 1 primers with one loop primer (LB), set 3 used the set 1 primers with another loop primer (LF), and set 4 used all six primers (Fig. 2). As expected, LAMP without the two loop primers showed the slowest amplification reaction. LAMP with one loop primer showed a faster amplification reaction than LAMP without any loop primer. LAMP with all six primers, including two loop primers, showed the fastest amplification reaction, in which a fluorescent signal appeared just 12 min after starting the reaction. None of the negative controls showed a positive signal during the reaction. Therefore, we decided to include all six primers in our Salmonella species LAMP system.

Optimization of the combinations of primers. Loop-mediated isothermal amplification (LAMP) reactions were performed with Salmonella enterica DNA with four different combinations of primers. (A) LAMP with two inner and two outer primers. (B) LAMP with the combination-A primers and one loop primer (LB). (C) LAMP with the combination-A primers and another loop primer (LF). (D) LAMP with all six primers. The x-axis represents the time needed for the LAMP reaction using a real-time PCR cycler; the y-axis represents the relative fluorescence signal. The flat amplification curve was found in the negative control.
Optimization of LAMP reaction conditions
To determine the best LAMP conditions, we compared three different reaction conditions: 60℃, 63℃, and 65℃ for 45 min (Fig. 3). Consistent with the results in Fig. 2, LAMP with all six primers including two loop primers showed the fastest amplification reaction regardless of reaction temperature. Regarding the reaction conditions, a reaction temperature of 60℃ showed the best amplification performance. Integrating these, we set the conditions for our Salmonella LAMP assay as using all six primers at 60℃ for 45 min.
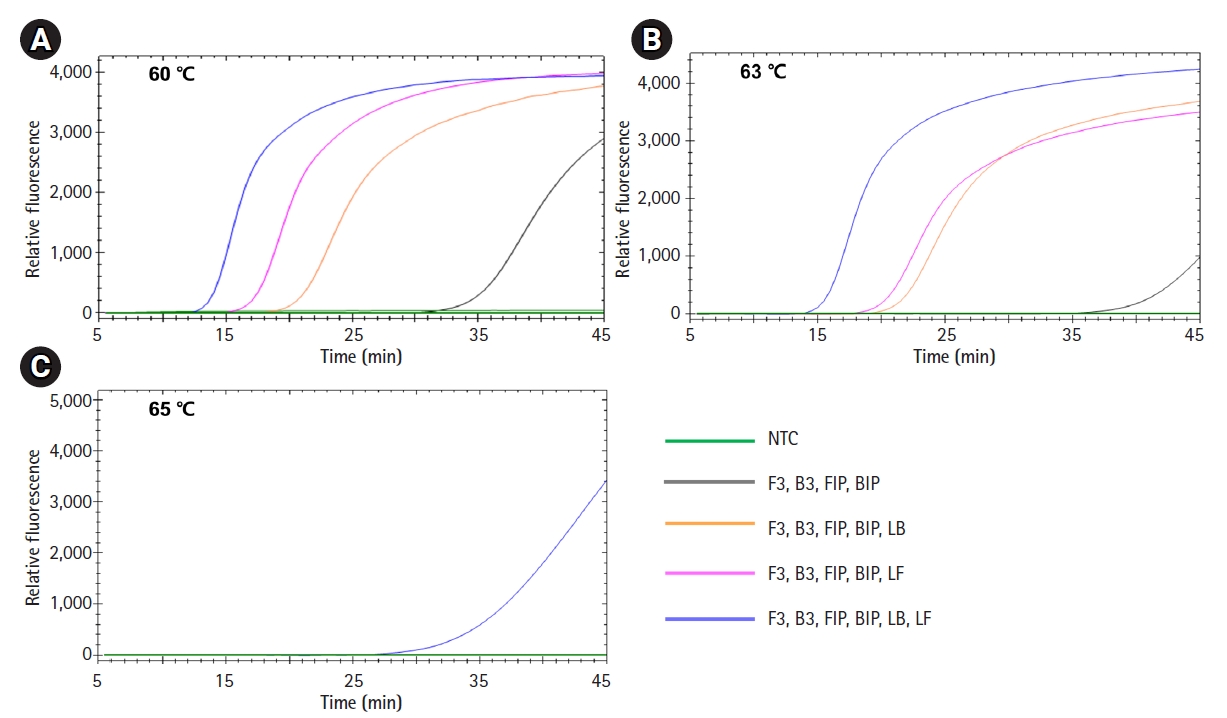
Optimization of loop-mediated isothermal amplification (LAMP) reaction conditions. LAMP reactions were performed with Salmonella enterica DNA under three different reaction conditions: 60℃ (A), 63℃ (B), and 65℃ (C). The combinations of primers were the same as shown in Fig. 2. NTC, negative control.
Detection sensitivity of the LAMP assay
To test the detection sensitivity of our Salmonella LAMP assay, we observed the limit of detection (LOD) of this assay. Salmonella enterica DNA was 10-fold serially diluted from 1 ng to 1 ag and used in the LAMP reaction (Fig. 4). The LOD of the Salmonella LAMP assay was 100 fg which corresponds to 20 copies of Salmonella DNA. The time for the fluorescent signal to appear from 1 ng of template DNA was less than 20 min. Even in the case of 1 pg of template DNA, the amplification signal appeared within 25 min.
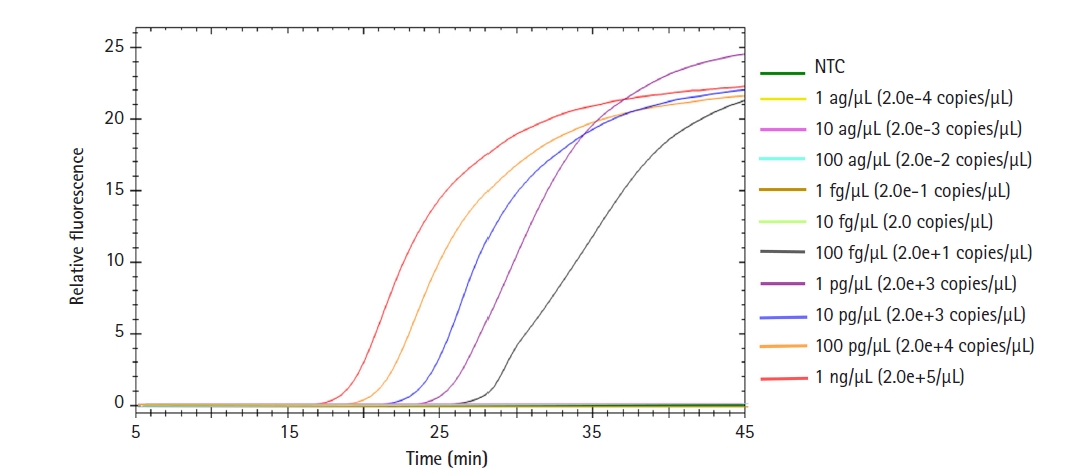
Sensitivity of the Salmonella species loop-mediated isothermal amplification (LAMP) assay. The template DNA (Salmonella enterica DNA) was 10-fold serially diluted (1 ng/μL to 1 ag/μL) and tested with our Salmonella LAMP assay. NTC, negative control.
Validating the universal detection of Salmonella species and specificity
To validate whether our universal Salmonella LAMP assay can detect diverse Salmonella species, we prepared seven clinically important Salmonella species (S. Typhi, S. Typhimurium, S. Enteritidis, S. enterica, S. Paratyphi A, S. Paratyphi B, S. Infantis) and used them with this assay (Fig. 5). All seven Salmonella species showed specific amplification signals at around 15 min except S. Paratyphi A. In contrast, the negative control did not show any amplification signal. To verify the Salmonella-specific detection, we also conducted our Salmonella LAMP assay with 11 non-Salmonella pathogens (seven Gram-negative and four Gram-positive bacteria) (Table 1). None of the non-Salmonella pathogens showed any amplification signal with our Salmonella LAMP assay (data not shown).

Universal detection of Salmonella species using our loop-mediated isothermal amplification (LAMP) assay. DNA extracted from seven clinically important Salmonella species (Salmonella Typhi, Salmonella Typhimurium, Salmonella Enteritidis, Salmonella enterica, Salmonella Paratyphi A, Salmonella Paratyphi B, Salmonella Infantis) was used in our Salmonella LAMP assay. NTC, negative control.
Discussion
In this study, to develop a real-time LAMP assay for the rapid and sensitive detection of Salmonella species, we designed a LAMP primer set targeting the hilA gene. The hilA gene, a member of the transcriptional regulator genes encoded in the Salmonella pathogenicity island 1 (SPI1), plays an important role in the pathogenesis of Salmonella infection by activating the expression of SPI1 [18,19]. SPI1 is one of the key virulence factors for Salmonella infection and invasion. The hilA gene is known to be Salmonella species-specific and absent in other Gram-negative bacteria [20]. Therefore, this gene has been targeted for detecting Salmonella infection by PCR [21]. However, no study has yet developing a LAMP assay targeting the hilA gene for the detection of Salmonella species.
In the LAMP assay, a high level of amplification efficiency can be achieved under isothermal conditions due to strand displacement by the Bst DNA polymerase enzyme [11]. However, the possibility of non-specific amplification is also high due to the high level of amplification. To minimize this possibility, we added NMF and IBA as described previously [17]. In addition, we optimized the LAMP reaction conditions to facilitate the efficient and reliable detection of Salmonella species.
When we checked the detection sensitivity of our Salmonella LAMP assay using S. enterica DNA, the LOD was 100 fg, which corresponds to just 20 copies of Salmonella DNA. In the previous PCR assay targeting the hilA gene using S. typhimurium DNA, the LOD was 100 pg [21]. Therefore, although the Salmonella strains used for the sensitivity test were different, our Salmonella LAMP assay is approximately 1,000 times more sensitive than the PCR-based assay to detect Salmonella species. Of note, when we tested whether our universal Salmonella LAMP assay can detect diverse Salmonella species, all seven Salmonella species (including S. Typhimurium) showed consistent detection performance, providing further support that our Salmonella LAMP assay is much more sensitive than the PCR assay. This result also suggests that this Salmonella LAMP assay is universally applicable for the detection of diverse clinically important Salmonella species.
When we tested the assay using 11 non-Salmonella pathogens, none of them showed any amplification signals with our Salmonella LAMP assay, suggesting high Salmonella species specificity of our LAMP assay. According to the BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) analysis, none of the 11 non-Salmonella pathogens showed similar sequences to the hilA gene (data not shown). Of particular note, a previous study based on the conventional PCR method targeting the hilA gene took approximately 1 hour and 45 minutes [21]. However, all Salmonella species tested in this study showed specific amplification signals at around 15 minutes except S. Paratyphi A, demonstrating that our Salmonella LAMP assay would be ideal for POCT.
One of the fundamental limitations of conventional PCR is that PCR is easily inhibited by substances in the sample. LAMP technology can overcome this drawback of PCR, as LAMP is relatively less sensitive to PCR inhibitors, which can contaminate samples from sources such as feces. Therefore, LAMP would be advantageous to minimize the sample preparation procedure.
In conclusion, our Salmonella LAMP assay targeting the hilA gene demonstrated a high level of sensitivity and specificity. Considering the higher sensitivity of our assay than conventional PCR, its rapid reaction time, and its lower sensitivity to potential PCR inhibitors, our assay could be a useful POCT tool for Salmonella species.
Notes
Authors’ Contribution
Conceptualization: JC, JS, YJC. Data curation: JC, JS, SS, YJC. Formal analysis: JC, JS. Funding acquisition: YJC. Methodology: JC, JS, SS, SK. Writing - original draft: JC. Writing - review & editing: YJC.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by a grant from the Korea Health Industry Development Institute (KHIDI) (HI21C0561) and a grant from the National Research Foundation of Korea (2017M3C9A6047615). We thank KREONET (Korea Research Environment Open NETwork) and KISTI (Korea Institute of Science and Technology Information) for allowing us to use their network infrastructure.