Somatic Mutaome Profile in Human Cancer Tissues
Article information
Abstract
Somatic mutation is a major cause of cancer progression and varied responses of tumors against anticancer agents. Thus, we must obtain and characterize genome-wide mutational profiles in individual cancer subtypes. The Cancer Genome Atlas database includes large amounts of sequencing and omics data generated from diverse human cancer tissues. In the present study, we integrated and analyzed the exome sequencing data from ~3,000 tissue samples and summarized the major mutant genes in each of the diverse cancer subtypes and stages. Mutations were observed in most human genes (~23,000 genes) with low frequency from an analysis of 11 major cancer subtypes. The majority of tissue samples harbored 20-80 different mutant genes, on average. Lung cancer samples showed a greater number of mutations in diverse genes than other cancer subtypes. Only a few genes were mutated with over 5% frequency in tissue samples. Interestingly, mutation frequency was generally similar between non-metastatic and metastastic samples in most cancer subtypes. Among the 12 major mutations, the TP53, USH2A, TTN, and MUC16 genes were found to be frequent in most cancer types, while BRAF, FRG1B, PBRM1, and VHL showed lineage-specific mutation patterns. The present study provides a useful resource to understand the broad spectrum of mutation frequencies in various cancer types.
Introduction
Recent progress in high-throughput sequencing technology has contributed to the generation of genome-wide somatic mutation profiles in diverse cancer samples. The Cancer Genome Atlas (TCGA) is one of largest collaborative efforts to generate multi-level omics data on human cancer tissue samples. Particularly, information on genome-wide somatic mutations has been collectively profiled from exome sequencing data from thousands of patients' tumor samples. Somatic mutation is a main driving force for cancer development and progression. Thus, many researchers have tried to complete the catalog of somatic mutations in cancer cell lines [1, 2]. Somatic mutation is also known to be involved in key mechanisms for cellular sensitivity or resistance against chemotherapy [3-5].
In our previous study using cancer cell line data, we reported that somatic mutation was a more significant classifier than cancer lineage in predicting the anticancer drug response [6]. Thus, we identified many unknown association patterns between cancer drug response and mutational genotypes in cancer cell lines-e.g., MYC-amp mutation-specific sensitivity of insulin-like growth factor 1 receptor inhibitors. In addition, mutation information provided important clues for us to better interpret the biological relevance of molecular signatures identified from the transcriptome and proteome data of diverse cancer cell lines. The next step should be to find out the clinical application of mutation-specific drug responses or molecular signatures obtained from cell line-based analysis.
Thus, it is important to systematically analyze the mutational genotype (mutaome) of various human tissue samples and identify mutations significantly associated with specific types of tumors. 'Mutaome' means the cancer mutational landscape, including mutations in oncogenes and tumor suppressors. In the present study, we organized all sequence-based mutation information into gene-based frequency data. Then, we comparatively determined the major genes of somatic mutations in diverse cancer subtypes and cancer stages (i.e., non-metastatic and metastatic samples). This work will provide practical information for directing in vitro cell line-based mutation-specific phenotypes to clinical applications in cancer drug discovery and mechanism studies.
Methods
Data acquisition
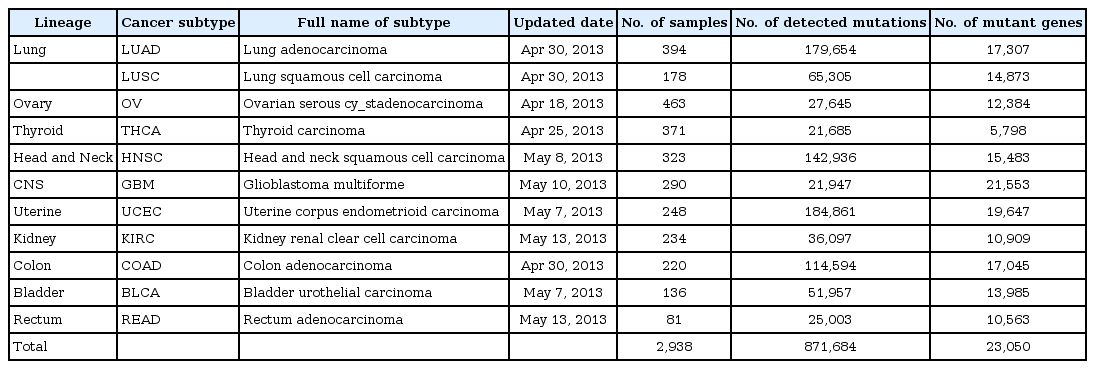
Somatic mutation data for tumor tissue samples from 2938 patients, harboring 11 cancer types, were obtained from the data portal of TCGA, which were freely available. These data (level 2) for 10 cancer types, except ovarian serous cy_stadenocarcinoma (OV), provide genome-wide somatic mutations on each sample experimented with the Illumina Genome Analyzer DNA Sequencing platform (Illumina, San Diego, CA, USA). The somatic mutation data for OV were organized, combined with data produced from Illumina and the ABI SOLiD DNA System Sequencing platforms (Applied Biosystems, Foster City, CA, USA). The details, including the full name of each cancer type and updated dates of mutation data, are provided in Table 1.
Survey of genome-wide somatic mutations in patient tumor samples
Together with somatic mutation data, clinical data for each patient were downloaded from the data portal of TCGA. These data were applied to categorize samples into metastasis and non-metastasis. The annotation of 'pathologic_M' was available to indicate the stage of metastasis in the patient's tumor samples. The stage of M0' means that there was no evidence of distant metastasis, and 'M1' means that a pathological distant metastasis was found. In this study, the samples from patients annotated as 'M0' were classified as non-metastatic, and those with 'M1' were classified as metastatic.
Analysis of somatic mutations
The number of detected mutations in each cancer type ranged from tens to hundreds of thousands. We organized the detected point mutations into 23,050 human genes. The number of mutant genes per sample was counted for ~3,000 samples. In addition, the mutation frequency and its percentage were calculated for all samples and each cancer type. The major mutant genes that ranked within the top 3 in each cancer type were selected based on the observed frequency in the overall, non-metastatic, and metastatic samples.
The patterns of frequency for the selected major genes were analyzed through hierarchical clustering method. The clustering and its visualization on a heatmap were performed using the software QCanvas [7]. QCanvas can be down-loaded freely from the website http://compbio.sookmyung.ac.kr/~qcanvas.
Results and Discussion
Mutation frequency in patients' tumor samples
From the TCGA database, thousands of patients' tumor samples were analyzed to detect variants in the whole genome. To quantify the genome-wide mutation profile in diverse tumors, we organized the whole mutant genes into several cancer types and stages, using the annotation information obtained from TCGA. Overall, a total of 871,684 mutations were virtually found in 23,050 human genes from 2,938 patient tumor samples (Table 1). Samples covered 11 diverse cancer types (Table 1). These data were continuously updated and produced by analyzing additional samples. Together with the extended production of multi-level omics data using the same patients' tissue samples, TCGA provides a useful resource for understanding the role of mutations in cancer progression.
The distribution of mutant genes on each sample showed that a variety of genes are mutated in individual tumor samples. Most samples contained on average 20-80 mutations (Fig. 1A). This means that each single tumor sample has mutations in multiple genes. Various mutations in a cancer sample were already well-characterized and constructed as open source data [8], and the significance of multiple mutations in a single tumor has been constantly suggested [9, 10]. It is appropriate that a tumor may also consist of a heterogeneous collection of cells with different types of mutations. Furthermore, in the aspect of lineage dependency, the amount of mutations in individual samples varied, depending on cancer type (Fig. 1C). Lung adenocarcinoma samples have a wide range of mutation frequencies in individual samples and harbor more mutations than other lineages. The broad range of mutation frequencies in lung cancer was also referred to in Lawrence et al. [11]. In contrast, thyroid carcinoma has relatively few mutations in individual samples. Further studies are required to understand the association with the amounts of mutations and major biological factors in each cancer type. This can be analyzed by comparing with the patient's clinical information, including smoking.
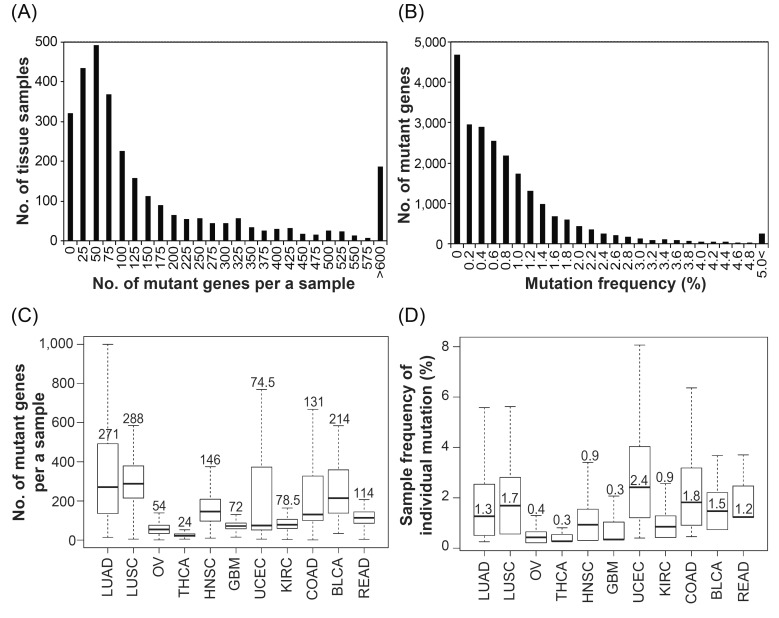
Overall mutation frequency in tissue samples. (A) The distribution of 2,938 patient tissue samples for the number of mutant genes per sample. (B) The distribution of 23,050 mutant genes for % mutation frequency. (C) Box plot of the number of mutant genes per sample for each caner subtype. (D) Box plot of % sample frequency of individual mutation for each caner subtype. Number in the box represents median value. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cy_stadenocarcinoma; THCA, thyroid carcinoma; HNSC, head and neck squamous cell carcinoma; GBM, glioblastoma multiforme; UCEC, uterine corpus endometrioid carcinoma; KIRC, kidney renal clear cell carcinoma; COAD, colon adenocarcinoma; BLCA, bladder urothelial carcinoma; READ, rectum adenocarcinoma.
On the other hand, the mutation of each gene was observed at a very low frequency in all samples (Fig. 1B). Most genes contained mutations in less than 1% of samples, and only a few genes contained mutations in over 5% of all samples. Generally, the mutation of a gene showed low frequency in all cancer types, except in uterine corpus endometrioid carcinoma (UCEC) (Fig. 1D). Mutant genes were found in 2.4% of UCEC samples (>2-fold greater than other lineages), and sometimes, a gene showed a mutation in >8% of UCEC tumors. In conclusion, there are only a few genes in which mutations are frequently (i.e., >1-2%) found in tumor. Genes with relatively frequent mutations in tumors may have a significant role in cancer progression.
Comparative analysis of mutations between non-metastatic and metastatic samples
TCGA provides annotations for the stage of metastasis in each patient sample from the clinical data. According to these data, thousands of samples for 8 cancer types, except for OV, glioblastoma multiforme (GBM), and UCEC, were classified into 89 metastatic and 1341 non-metastatic samples in order to compare the mutation frequency between them. The annotation for metastasis was not provided for the excluded cancer types-OV, GBM, and UCEC. Interestingly, metastatic samples had similar mutation frequencies as non-metastatic samples for most cancer types (Fig. 2). Kidney renal clear cell carcinoma (KIRC) had significant (p < 0.01) difference in frequency between metastatic and non-metastatic samples. This result implied that there is no differential occurrence of mutations between metastatic and non-metastatic samples, except in minor case.
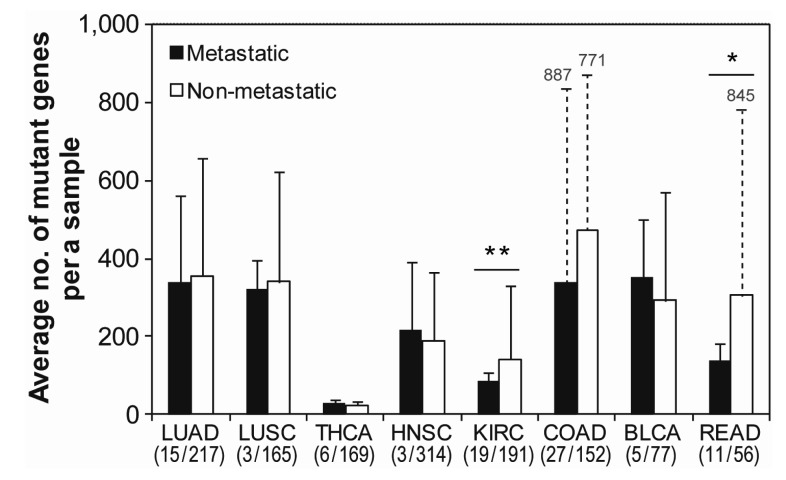
Comparison of mutation frequency between metastatic and non-metastatic cancer samples. Three cancer types-OV, GBM, and UCEC-were excluded, because the information on metastasis was not provided. The lines on the bar represent the standard deviation for the number of mutant genes per sample. The numbers on the dotted lines represent the value of the standard deviation in each sample. The significant difference between metastatic and nonmetastatic samples was considered based on the probability of t-test. **p < 0.01 and *p < 0.1. The numbers under the x-axis show the number of metastatic and non-metastatic samples in each cancer type. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cy_stadenocarcinoma; THCA, thyroid carcinoma; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; COAD, colon adenocarcinoma; BLCA, bladder urothelial carcinoma; READ, rectum adenocarcinoma.
Identification of major mutant genes
In this study, the mutation frequency was analyzed separately in overall, non-metastatic, and metastatic samples. We focused on mutant genes exhibiting high frequency in each sample group. A total of 12, 10, and 15 genes were ranked within the top 3 mutations in at least one of cancer type in the overall, non-metastatic, and metastatic samples, respectively (Table 2). Especially, TP53 and TTN showed dominant frequencies for over 1,000 of all samples.
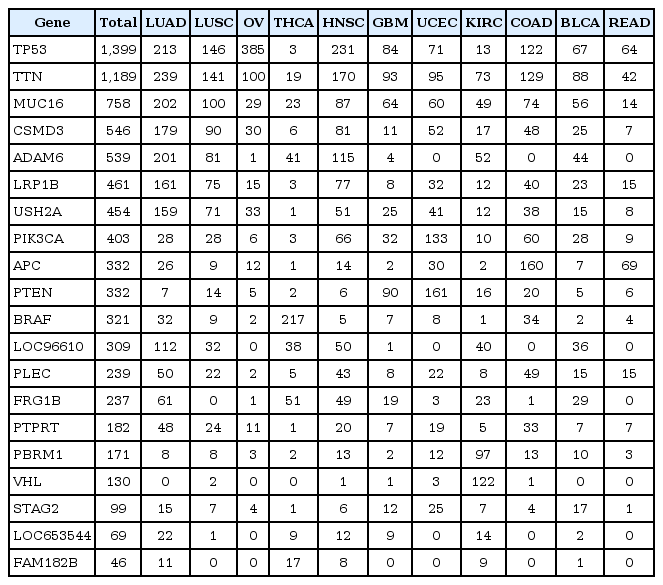
List of mutant genes showing high mutation frequency in each cancer lineage
In addition, the pattern of mutation frequency for the selected major mutant genes was analyzed in diverse cancer categories (Fig. 3). Regardless of sample group, MUC16, TTN, and TP53 were found to be frequent in most cancer types. TP53 is a well-known mutant gene, playing an important role in cancer progression [12, 13]. It was reported that TP53 mutation is frequently represented in major cancer lineages [6]. Mutation TTN and MUC16 has not been reported to be critical in cancers. This analysis shows that they may have potential, specific roles in cancer development or progression.
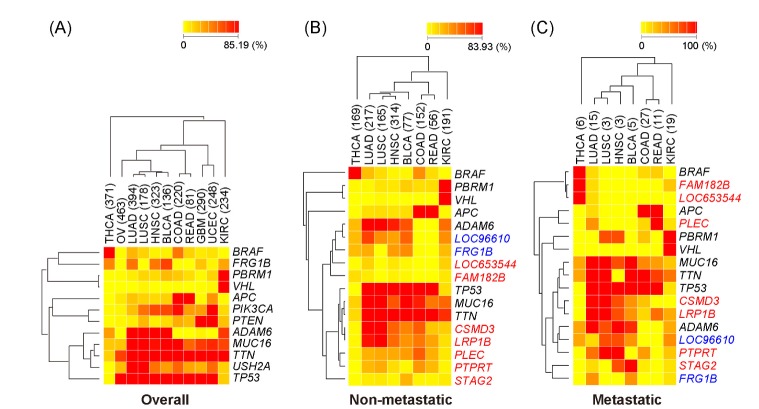
Lineage-dependent frequency of major mutant genes. (A) Heatmap of the 12 major mutant genes in overall samples. Twelve mutant genes are ranked in the top 3 in at least one cancer type based on the % frequency in the overall samples. (B) Heatmap of the 17 major mutant genes in non-metastatic samples, ranked in the top 3 in at least one cancer type based on the % frequency in metastatic and non-metastatic samples. Two genes colored in blue are derived from non-metastatic samples, and 7 genes colored in red are derived from metastatic samples. Others colored in black are commonly selected in metastatic and non-metastatic samples. (C) Heatmap of the 17 major mutant genes in metastatic samples. The list of genes is the same as in Fig. 3B. Red represents high % frequency and yellow represents low % frequency. In the heatmap for metastatic and non-metastatic samples, three cancer types-OV, GBM, and UCEC-were excluded, because the information on metastasis was not provided. THCA, thyroid carcinoma; OV, ovarian serous cy_stadenocarcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; HNSC, head and neck squamous cell carcinoma; BLCA, bladder urothelial carcinoma; COAD, colon adenocarcinoma; READ, rectum adenocarcinoma; GBM, glioblastoma multiforme; UCEC, uterine corpus endometrioid carcinoma; KIRC, kidney renal clear cell carcinoma.
Among 12 major mutant genes selected from the overall samples, BRAF, FRG1B, PBRM1, and VHL had cancer subtype-specific mutation patterns (Fig. 3A). Especially, PBRM1 and VHL showed strong specificity in KIRC tumors. As previously reported [14], the alterations of VHL (a tumor suppressor gene) are clearly dominant in renal cell carcinoma. Together with VHL, PBRM1 was identified as a major gene, frequently mutated in renal carcinoma [15]. An association with the loss of its expression and renal cell carcinoma progression was suggested in previous studies [16].
Mutant genes dependent on metastasis
We found that there was no difference in the overall frequency of metastatic and non-metastatic samples (Fig. 2). The nine mutant genes found in non-metastatic samples were all included in the major mutant genes in the overall samples (Fig. 3B). The lineage-dependent frequency in non-metastatic samples was also similar with the overall pattern in Fig. 3A. However, half of the 15 major mutant genes from metastatic samples were different from the mutant genes in non-metastatic or overall samples (Fig. 3C). The mutations of FAM182B, LOC653544, PLEC, STAG2, PTPRT, CSMD3, and LRP1B represented unique frequencies in metastatic samples. Especially, PTPRT is a member of the protein tyrosine phosphatase (PTP) family. The deletion of PTPRD, included in the same PTP family, is frequently seen in metastatic cutaneous squamous cell carcinoma [17]. Further studies are required for other major metastasis-associated mutant genes. In conclusion, the diversity of major mutant genes in metastatic samples is quite different from those in non-metastatic tumors, although the overall mutation frequency is similar between metastatic and non-metastatic tumors. The present study provides a useful resource for understanding the varied frequency of diverse mutations in patients' tumor samples.
Acknowledgments
This research was supported by Sookmyung Women's University Research Grant 1-1203-0227.