A Primer for Disease Gene Prioritization Using Next-Generation Sequencing Data
Article information
Abstract
High-throughput next-generation sequencing (NGS) technology produces a tremendous amount of raw sequence data. The challenges for researchers are to process the raw data, to map the sequences to genome, to discover variants that are different from the reference genome, and to prioritize/rank the variants for the question of interest. The recent development of many computational algorithms and programs has vastly improved the ability to translate sequence data into valuable information for disease gene identification. However, the NGS data analysis is complex and could be overwhelming for researchers who are not familiar with the process. Here, we outline the analysis pipeline and describe some of the most commonly used principles and tools for analyzing NGS data for disease gene identification.
Introduction
The breakthrough in next-generation sequencing (NGS) techniques has enabled rapid sequencing of whole genomes at low cost and has brought tremendous opportunities as well as challenges to biomedical research [1-5]. The cost of sequencing is projected to continue to drop, with whole-genome sequencing expected to be as low as $1000 [4] and whole-exome sequencing (i.e., sequencing approximately 1% of the coding regions of the genome [6]) to be $500 [1]. Such low costs enable a small research lab to generate large amounts of sequence data in a short period of time on a relatively small budget. The low cost also allows sequencing-based clinical genetic tests to be routinely performed to provide guidance for health professionals for disease etiology and management. The challenge now is how to reliably synthesize and interpret this large amount of raw data from sequencing platforms.
One goal of genomic research is to determine which genes are causal for the disease of interest. With the NGS data, this is not a trivial task. Large-scale whole-genome and whole-exome sequencing analyses identify large amounts of genomic variants, most of which are not related to disease risk. Thus, disease gene prioritization principles and tools are needed [2] to identify and prioritize variants of interest from the large pool of candidates. Currently, identifying candidate causal mutations/genes is often a complex practice that involves several dozens of steps and/or tools to accomplish. These tools work jointly in a series of steps and thus functionally form a "workflow" or a "pipeline." Ideally, the process of disease gene prioritization generates an ordered list of genes that puts high-risk candidates on the top of the stack and filters out "benign" or neutral variants. In reality, this process usually involves multiple filtering steps, some of which have underlying assumptions that can affect how variants are filtered.
The focus of this review is to provide a broad overview of the workflow and present some of the bioinformatics software tools that are currently available. More in-depth comparisons of the bioinformatics algorithms and tools are described elsewhere [3, 5].
Workflow for Disease Gene Filtering/Prioritization
Typically, the output from NGS platforms is in the standard FASTQ format. The FASTQ file is a text file containing a list of short DNA fragments, generally less than 150 base pairs (bp) long, and a list of quality scores associated with the DNA fragments. For whole-genome or whole-exome sequencing, a FASTQ file contains millions of records. For example, the human exome is ~50 million base pairs (Mbp), and a typical whole-exome sequencing dataset has at least 30X depth of coverage (i.e., each bp is sequenced 30 times on average). Such a FASTQ file will contain ~15 million raw 100-bp sequences. To identify the causal genes from this raw data, a variant gene prioritization pipeline usually involves three stages or phases of data processing: 1) raw sequence processing and mapping, 2) variant discovery and genotyping, and 3) disease gene filtering and/or prioritization.
Phase 1: Raw sequence processing and mapping
Overview
The goal of Phase 1 is to preprocess the FASTQ file and evaluate the quality of the raw reads and then to map or align the sequences to the reference genome-that is, to find the best location on the reference genome where each sequence might have originated. Successfully mapped sequences are annotated with information that includes the location on a specific chromosome and how well the sequence matches the reference genome. After initial mapping, there are several subsequent recalibration steps to improve the mapping results.
Preprocessing
The raw FASTQ file contains all of the raw sequences, some of which are of low quality. The overall quality should be evaluated, and the lower-quality reads should be removed before mapping to the reference genome to improve mapping efficiency. FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) is a popular tool that produces quality analysis reports on FASTQ files. FASTQC provides graphical reports on several useful statistics, such as "Per base sequence quality," "Sequence duplication levels," and "Overrepresented sequences," etc. Specifically, "Per base sequence quality" determines if trimming low-quality sequences is needed before the mapping/alignment step; "Sequence duplication levels" evaluates the library enrichment and complexity; and "Overrepresented sequences" evaluates potential adaptor contamination. Low-quality bases and adaptor contaminations can cause an otherwise mappable sequence not to map and therefore should be removed (e.g., trim the 3' end of the read. which tends to have low quality). FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) provides a set of command line tools for manipulating FASTQ files, such as trimming input sequences down to a fixed length or trimming low-quality bases. Cutadapt (https://github.com/marcelm/cutadapt) can be used for trimming short reads to remove potential adapter contamination.
Mapping
Once high-quality sequence data are obtained, a number of bioinformatics programs, including Mosaik [7], MAQ (http://maq.sourceforge.net/), mrFAST [8], BWA [9], Bowtie2 [10], SOAP2 [11], and Subread [12], can be used to perform short-read sequence alignment to the reference genome. Several recent studies provided a detailed description and comparison of the alignment programs [5, 13], although it could be difficult to choose among aligners, because benchmark results vary across studies [10, 12, 14]. BWA and Bowtie2 are two popular programs for alignment. Both programs employ the Burrows-Wheeler transform (BWT) algorithm and have been shown to yield very good overall performance [13]. Bowtie2 is often used for mapping ChIP-seq and RNA-seq data, and BWA is often used for mapping whole-genome/exome data. We highlight the BWA program here, because it is accurate, fast, thoroughly tested, and well supported. Additionally, BWA has been utilized in multiple large-scale projects and well-defined workflows, including the Genome Analysis Toolkit (GATK) by Broad Institute [15], the 1000 Genome Project [16], the NHLBI GO Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/), and the HugeSeq workflow [17]. Various commercial companies, such as Seven Bridges Genomics (https://www.sbgenomics.com/) and Geospiza GeneSifter (http://www.geospiza.com/), also utilize BWA in their standard workflows.
After mapping the reads, the final output file from most software is typically in the sequence alignment/map (SAM) format [18]. In some cases, the SAM file is converted to its binary format (BAM) using SAMtools [18] to reduce the file size and optimize computation performance.
Recalibration
After the initial mapping procedure, subsequent recalibration steps are typically employed to improve the mapping results, such as performing local realignment or de novo assembly for regions that contain insertions/deletions (indels), removing PCR duplicates, and recalibrating the base quality scores. Indel realignment is a highly recommended post-BAM processing procedure. Due to the trade-off between speed and accuracy, indels are not well aligned by most general purpose aligners. This could lead to false variant calls in downstream analyses. Regions with high probability of potential indels can be realigned locally using IndelRealigner, part of the GATK toolkit. Another commonly used recalibration procedure is removing PCR duplicates. If a DNA fragment is amplified many times by PCR during the sequencing library construction, these artificially duplicated sequences can be considered as support of a variant by downstream variant discovery programs. Some BAM processing programs, such as Picard (http://picard.sourceforge.net/) and Samtools [18], can identify these artificially duplicated sequences and remove them. Base recalibration is also a recommended step, because the sequencer may have assigned a biased quality score upon reading a base (e.g., the score of a second "A" base after a first "A" base may always receive a biased quality score from a sequence machine [19]). Tools, such as Base-Recalibrator in the GATK toolkit, can calibrate the quality score to more accurately reflect the probability of a base mismatching the reference genome. One additional optional step, recommended by GATK, is data compression and reads reduction, especially for high-coverage data. For example, if a large chunk of sequences matches the reference exactly, it is not necessary to keep all the data, as they do not carry useful information for downstream analyses (assuming we are only interested in the sites that are different from the reference genome). In such a scenario, keeping one copy of each of the consensus sequences may be sufficient, and the redundancies can be removed to reduce file size and enable faster downstream computing. However, keeping a copy of the original file is highly recommended after data compression.
Phase 2: Variant discovery and genotyping
Overview
In many scenarios, only the sites that differ from the reference genome are of interest, because sites that are identical to the reference genome are not expected to be related to pathological conditions. Once raw sequences are properly mapped to the reference genome, the next step is to find all positions in an individual's genome that differed from the reference. This phase is referred to as variant discovery, or variant calling. Similar to the mapping phase, variant calling also contains an initial discovery step, followed by several filtering processes to remove sequencing errors and other types of false discoveries, and finally, the individual genotypes are inferred (i.e., if a locus is heterozygous, homozygous, or hemizygous for the variant). The output of variant calling contains all the variants and related information. Sites that are identical to the reference genome (i.e., invariant sites) are usually not included in the output variant file.
Variant discovery and genotyping
A number of variant calling software packages can be used to identify variants and call individual genotypes. Some of the commonly used software programs are SAMtools [18], freebayes (http://github.com/ekg/freebayes), SNPtools [20], GATK UnifiedGenotyper, and GATK HaplotypeCaller. Some of the tools, including SAMtools, SNPtools, and the GATK UnifiedGenotyper, use a mapping-based approach. Other tools, such as freebayes and the GATK Haplotype-Caller, use a local assembly approach. A more detailed survey and comparison of the tools have been previously described [5, 21]. These procedures typically take the BAM files from the "raw sequence processing and mapping" phase, together with the reference genome sequence in a FASTA file. The output file from this step is usually in the standard variant call format (VCF). Some of the tools allow the user to specify known variants to optimize the variant discovery. For example, GATK UnifiedGenotyper uses known variants (e.g., from the dbSNP database) as high-confidence datasets to assist model training. After variant and genotype calling, the probability scores of variants are recalibrated, and artifacts are identified. The artifacts can be removed subsequently either by filtering the data using a series of criteria or by building and applying an error model.
One example of the variant discovery tool is the GATK UnifiedGenotyper, which calls variants by examining the reference genome base by base, without correcting misalignment issues. This procedure could lead to false positive calls, particularly in regions containing indels. To increase the accuracy and sensitivity, calling multiple samples simultaneously is recommended. By taking into account the information of a variant in a population, the accuracy and sensitivity of variant discovery are greatly improved, especially for common variants [16, 22]. The GATK Haplotype-Caller, as compared to the UnifiedGenotyper, calls all variants (single nucleotide polymorphisms [SNPs], indels, structural variations, etc.) simultaneously by performing local de novo assembly of haplotypes and emits more accurate call sets, with the drawback of being slower. In general, structural variations (SVs) and copy number variations (CNVs) are more difficult to detect than SNPs and indels because of their heterogeneous nature. For SVs and CNVs, it is generally recommended to apply a combination of several tools and take the overlapping variant sites for high-confidence calls [17].
Genotype phasing and refinement
After the initial variant discovery and recalibration, variant genotyping and/or phasing are performed to provide critical information for downstream medical and population genetic studies that require accurate haplotype structures. For example, Mendelian diseases may be caused by a compound heterozygote event where heterogeneous mutations are found on different chromosomes. In such a scenario, knowledge about the paternal or maternal origin of a variant can provide valuable information. The GATK workflow recommends three steps of genotype phasing: transmission based on pedigree, haplotype based on reads, and statistical imputation. GATK PhaseByTransmission incorporates pedigree data to assist genotype calling, while GATK Read-BackedPhasing performs physical phasing of variant calling based on sequence reads. Statistical imputation of genotypes can be processed by several programs, such as Beagle [23,24], IMPUTE2 [25], and MACH [26].
Phase 3: Disease gene filtering and/or prioritization
Overview
A typical variant call pipeline will produce approximately 3 million variants from one whole-genome sequencing data set and more than 20 thousand variants from one whole-exome sequencing data set [27]. Because disease causing mutations might just be several "needles" in this tremendous "haystack" of variants, rigorous algorithms and high-quality databases are essential to accurately locate the candidate genes. This process is done either by filtering out variants that are not likely to carry a disease risk or by ranking all variants based on biological/statistical models, such that high-risk candidates rank at the top of the list [1, 2, 4]. Unlike Phase 1 and Phase 2, which have largely converged to well-formed computation workflows and platform-free file formats (e.g., FASTQ, BAM, and VCF formats), Phase 3 has more variations in the tools and workflows to suit the different needs of various research or clinical projects.
Annotation
Although many different strategies can be used to search for disease-causing mutations, some general procedures are shared. The first step, variant annotation, typically investigates the potential pathogenic impact of a variant, before any subsequent filtering or prioritizing. The goal is to identify and report the effect of each variant with respect to predefined "features," such as genes. For example, if a variant (SNP or indel) is present within the coding region, the potential effects include change to amino acid (non-synonymous), no change to amino acid (synonymous), introduction of stop codon (stop gain), removal of stop codon (stop loss), addition/removal of amino-acid(s) (inframe indel), and change to the open reading frame (frame-shifting indel), etc. Other than variant call files, the annotation process requires a feature file that defines the location of genomic features (e.g., transcript, exon, intron, etc.), which is usually in browser extensible data (BED) format [28], or generic feature format version 3 (GFF3) format maintained by the Sequence Ontology project [29]. In many situations, the reference genome in FASTA format is also provided as part of the input, and the annotated variants are likely to be platform-specific [30, 31]. Annotation is part of the workflow for almost all variant analysis tools, such as the open source software package VAAST [31], ANNOVAR [27], and many commercial service providers (e.g., Ingenuity, Golden Helix, Geospiza GeneSifter, Omicia, Seven Bridges Genomics, Biobase, etc.).
Once the variants are annotated, a number of methods can be used to predict the severity of a variant, based either on the protein sequence conservation information (e.g., BLOSOM62, SIFT) or the DNA sequence conservation information (e.g., PhastCons). The SIFT [32] score system assumes that protein "motifs" or "sites" are functionally important if they are highly conserved across species. For example, coding regions, active sites of enzymes, many splice sites, and promoters are usually conserved across species [32]. Technically, SIFT uses protein homology to calculate position-specific scores, which are then used to evaluate if an amino acid substitution at a specific location is damaging or tolerated. The accuracy of the SIFT prediction depends largely on the availability and the diversity of the homologous sequences across species. Therefore, the application and accuracy of SIFT are limited if there is limited homolog information or if the diversity is low. The BLOSUM62 matrix [33] is a more general-purpose score system that is based on the alignment of protein homologs with a maximum of 62% identity. The BLOSUM62 matrix is then calculated based on the count of the observed frequency of specific amino acid substitutions. The PhastCons score [34] is calculated based on DNA sequence conservation in a multiple alignment. It uses a statistical model of sequence evolution and considers the phylogeny relationship of multiple species. Instead of measuring the statistical similarity and diversity in percent identity, PhastCons uses a phylogenetic Hidden Markov Model and provides base-by-base conservation scores. Because the PhastCons score is calculated for each base, it can be applied to regions beyond the coding sequence to provide valuable information for prioritizing non-coding variants.
In addition to the conservation information, many software packages also incorporate protein structure information, such as the positions of active sites and secondary structures, into the prediction algorithm (e.g., Polyphen2, SNAP). Polyphen2 [35] combines eight sequencing features (e.g., congruency of the variant allele to the multiple alignment) and three structural features (e.g., changes in hydrophobic propensity) to score an amino acid substitution. The homology search and model training of Polyphen2 are based on the UniProt database. It worth noting that Polyphen2 provides two different prediction models that use different datasets for training, with HumVar tuned to score Mendelian diseases and HumDiv tuned to evaluate rare alleles at loci that are potentially involved in complex traits. SNAP [36] is another tool that uses various protein information, such as the secondary structure and conservation, to predict the functional effect of a variant.
Filtering
After variant annotation, two types of disease-gene identification strategies are commonly employed. A filter-based approach uses a series of criteria to sort out variants or genes that are unlikely to be causative, given a disease model. For example, in a typical workflow of ANNOVAR, variants that overlap with the 1000 Genome Project are removed from the list. Since the 1000 Genome Project aimed to identify common SNPs with a minor allele frequency (MAF) greater than 1%, these variants are thought to be "benign" in a rare Mendelian disease, under the assumption that the causing mutation of a rare disease should have a very low MAF. The HapMap [37, 38] database, which intended to identify haplotype-defining variants in different populations, can also be used for variant selection. In some cases, high frequency of a haplotype can be related to a specific disease, and can help researchers focus on a subset of variants within a specific region. Another commonly applied filter is to exclude non-coding variants under the assumption that a disease-causing mutation should occur in the coding regions and affect protein sequences. Several other filtering criteria are also commonly used, such as filtering variants with little predicted functional impact (e.g., synonymous variants) or having low-quality scores (e.g., low read-depth or low genotype quality score). A disease model can also help filter variants. For example, a disease under a recessive mode of inheritance requires more than one deleterious variant in a gene.
The filtering strategy is intuitive and easy to perform and has been successful in early whole-genome/exome studies on rare, single-gene Mendelian diseases. Numerous tools have been developed to apply various filtering strategies (reviewed in [1, 2, 4, 5]). However, caution should be taken when studying common, complex diseases, because applying hard filters could remove real casual variants. For instance, common diseases could be caused by common variants that have incomplete penetrance (i.e., not all individuals carrying the variant will have the disease).
Ranking
To overcome the difficulties associated with hard filtering, a prioritizing approach ranks each gene or feature based on the cumulative disease risk of potential deleterious variants. As an example, the VAAST tool kit performs the composite likelihood ratio test to determine the risk of a gene or a feature [30, 31]. Information of each variant within the gene, such as the predicted biological impact, allele frequency, and the conservation score of the variant allele, is considered simultaneously under a likelihood framework. One advantage is that it reduces the risk of filtering out potential risky coding variants, since there are no hard filtering steps. Another advantage is that it can score all variants, including non-coding variants that would have otherwise been filtered out. The statistical framework used by VAAST considers two sets of information: 1) the likelihood of observing the MAF of a variant under a disease model versus a non-disease model; and 2) the likelihood that a variant is deleterious versus non-deleterious. A combination of the observed frequency and predicted functional impact of variants is used to assess the disease risk of a gene.
A practical consideration is that most variants in the sequencing study are rare and may be difficult to achieve sufficient power for a statistical model [39]. To overcome this, a common solution is to assess the cumulative effects of multiple variants in a defined genomic region, such as a gene or an exon [39-41]. This approach evaluates the overall genetic burden of multiple rare variants and is referred to as the burden test. Two general methods are commonly used to collapse multiple rare variants [40], known as the cohort allelic sum test (CAST) and combined multivariate and collapsing (CMC) method. The CAST method measures the differences (between case group and control group) in the number of individuals who carry one or more rare variants [39, 42], while the CMC method treats all rare variants as a single count for analysis with common variants [39]. Since both methods implicitly assume that all variants influence the phenotype in the same direction [39, 40], it could introduce substantial noise if a lot of the rare variants are neutral [40]. Alternatives to these two basic methods are also available [40]. For example, by default, the VAAST program combines all variants that have less than three copies in a gene into one pseudo-site for scoring.
Pathway analysis
Pathway analysis is an alternative way to search for deleterious genes. The assumption is that a single mutation in a single gene may not be very harmful but that a combination of mutations in several genes causes the disease. In this type of analysis, variants are put in a context of biological processes, pathways, and networks to gain global perspective on the data. A large number of knowledge bases and tools have been developed for this task (reviewed in [43, 44]). In general, pathway systems contain a curation process that is either manual or automatic, followed by database assembly and refinement [43]. Many public knowledge base and analysis tools are available, such as Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.kegg.jp/); BioCyc (http://www.biocyc.org/), provided by SRI international; PANTHER [45]; Reactome [46]; and DAVID [47]. Optionally, commercial systems, such as Ingenuity Pathway Analysis (http://www.ingenuity.com/products/ipa), Pathway Studio (http://www.elsevier.com/online-tools/pathway-studio), and GeneGo (http://portal.genego.com/), are also available. Ingenuity Variant Analysis (http://www.ingenuity.com/products/variant-analysis) is also backed up with Ingenuity Knowledge Base and can consider the upstream and downstream genes of a variant in a pathway and evaluate the potential role of a gene by incorporating the related genes.
Gene Identification Pipeline: An Example
To help readers who have limited experience with NGS data to understand the workflow, in this section we will show a real example of a pipeline that was used to process whole-exome sequencing data from raw sequences to a ranked candidate gene list (Fig. 1). The pipeline is constructed, based on the three-phase organization, as described in the previous sections. We start from raw sequence FASTQ files and assume no quality control has been performed or reported. Please note that each tool is likely to require dependent files, such as the reference genome, the 1000 Genome variants, dbSNP variants, and the genome feature file in GFF3 format, etc.
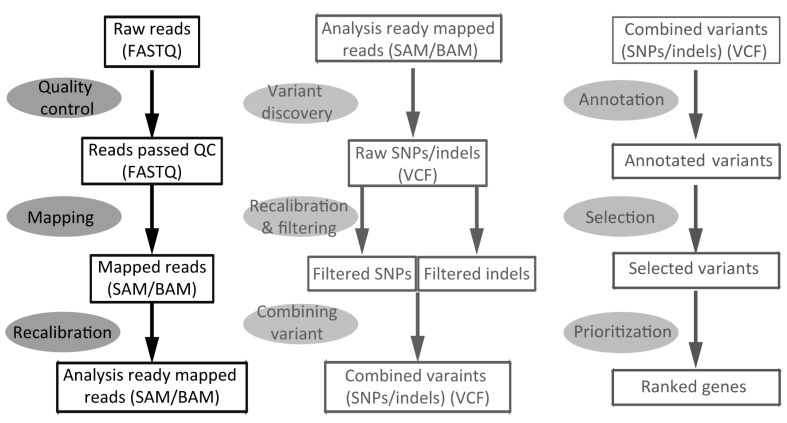
Example workflow for disease gene identification using next-generation sequencing data. SAM, sequence alignment/map; BAM, binary format; SNP, single nucleotide polymorphism; VCF, variant call format.
Sequence mapping
- Raw sequence quality control. FastQC can be used to perform graphical quality checks on the raw sequence. FastX can be used to filter the reads by specifying quality score ranges.
- Initial mapping. BWA is recommended for mapping Illumina reads. The first step of BWA is to generate suffix array coordinates for mapping the reads; then, one performs the actual alignment and outputs SAM format files. The SAM file is converted to BAM format, sorted, and indexed using Samtools for faster downstream processing (sorting is often required by many downstream tools).
- Alignment recalibration. The GATK RealignerTarget-Creator tool is used to create target regions for realignment, followed by the IndelRealigner tool to perform the actual realignment and output new BAMs. The Picard MarkDuplicates tool is then used to mark and remove duplicated sequences, followed by the Picard BuildBamIndex tool to rebuild the index. Next, GATK's BaseRecalibrator tool is used to recalibrate the base quality scores, and PrintReads is used to output the final, analysis-ready reads in BAM format.
Variant discovery
- Initial variant discovery. The GATK UnifiedGenotyper tool is used to perform initial raw variant calling and output VCF files.
- Variant recalibration and filtering. SNPs and indels are processed separately but in a similar manner; so, we will only describe SNPs as an example. The GATK Select-Variants tool is used to select SNPs within the exome regions from raw VCF files. The VariantRecalibrator tool is then used to build Gaussian models for recalibration, followed by the ApplyRecalibration tool to apply models for SNP recalibration. Low-quality SNPs are then removed by imposing a filtering step, based on a user-defined variant quality score recalibration (VQSR) score threshold. After SNPs and indels are reprocessed, the GATK CombineVariants tool is used to combine the recalibrated SNPs and indels.
Disease gene prioritization
- Variant annotation. VCF files are converted to genome variation format (GVF) format using the VAAST vaast_converter tool. The variants are then annotated using the VAAST VAT tool.
- Variant selection. The annotated variants are selected and condensed to condenser (CDR) format using the VAAST VST tool for cases and controls, respectively.
- Gene prioritization. VAAST is used to perform disease gene ranking and output a text file containing a list of ranked candidate genes.
Automated Workflows vs. Flexible Pipelines
High-throughput data processing and analysis are becoming reliable and efficient, thanks to numerous high-quality tools that are mostly open source, with long-term active support and frequent updates to incorporate new advances in the field. A typical workflow covering all three phases outlined above involves many different tools and/or steps, databases, score systems, and file re-formatting steps. Therefore, it is not a trivial task to perform a data processing task, starting from the raw FASTQ files to the identification of candidate genes. There are many tools and dependencies of tools that need to be installed and many steps and many parameters that need to be specified. This could be a big hurdle for researchers that have limited experience with NGS data pipelines. It would be a good idea for the community to converge on several standardized, well-configured, project-optimized, and automated workflows that can be used widely with minimum user interface. HugeSeq [17] by Stanford is a comprehensive, automated workflow that integrates around two dozen bioinformatics tools and can process FASTQ/FASTA files, perform various quality control and clean-ups, and output variant call files. Commercial services, such as Seven Bridges Genomics and Geospiza, also have preconfigured, comprehensive, automated pipelines for their customers.
Flexibility of pipelines, on the other hand, may be important for researchers who want to have complete control on the pipeline design to fit their projects. In such cases, it is important to have plasticity for users to customize pipelines. For example, a flexible pipeline system should allow users to integrate specific tools that are not part of a general purpose workflow and allow users to supply user-defined control files (e.g., providing a BED file to restrict the variant call within specific regions). There are a number of such pipelines that are well defined and easy to use, such as the GATK pipeline, gkno system (http://gkno.me/), Galaxy [48-50], and VAAST [30, 31].
Conclusion and Future Direction
Highly efficient and accurate bioinformatics tools are available for most of the steps for analyzing sequence data, from processing the raw NGS data to generating the final report of potential risk genes. Users can choose automated workflow versus customized pipelines to suit their research projects. Many bioinformatics tools are optimized, such that a small server (e.g., 48 cores, 250 GB RAM, 2 TB storage) can perform the whole analysis workflow within a reasonable time (e.g., 250 hours in nonparallel mode and 25 hours in parallel mode for one exome at 30× coverage, as benchmarked by HugeSeq). Furthermore, the scientific community is converging towards standard workflows and standard file formats, crossing different research institutes, companies, and platforms. Several file formats are becoming standard, such as FASTQ/FASTA, SAM, BAM, VCF, and GFF3. Large publically funded projects, such as the 1000 Genome Project, the HapMap Project, and the NHLBI GO Exome Sequencing Project, have played important roles in the unification process.
Nevertheless, many challenges remain, and we would like to highlight a few: 1) data standards still need wider agreement. For example, a number of variant discovery tools for SV and CNV use different file formats and need to be converted to the more standard format for downstream analysis [17]; 2) more standardized, automated workflows need to be developed to accommodate different data and projects and minimize the user interaction, such that different research groups can perform independent data analysis and the results can be easily compared; and 3) large, high-quality, and unified databases are essential for disease gene identification/prioritization, and continuous efforts from the scientific community are needed.
In addition to the challenges, a number of improvements that are urgently needed for current analysis pipeline are being actively developed and implemented:
- Sequence mapping. Apply haplotype-based mapping and de novo assembly to reduce mismatches and increasing specificity.
- Variant discovery and genotyping. Improve the sequencing technology (e.g., longer read length) and analytical methods (e.g., de novo assembly-based variant discovery) to identify and infer genotypes of complex variations, such as SVs, CNVs, large indels, and transposons.
- Candidate gene prioritization. Develop complex prioritization strategies for large pedigrees, combine linkage analysis with association studies, and integrate pathway analysis to eliminate false positive genes.
- Non-coding variant annotation. Understand the functional impact of non-coding DNA elements, as the Encyclopedia of DNA Elements (ENCODE) Project intended to do, to greatly broaden our view of disease-causing mutations.
- Cloud computing. High-throughput sequencing projects generate large amounts of data, create huge computational challenges, and require numerous tools and libraries for comprehensive data analysis. Develop cloud-based computing resources, such as the 1000 Genome Project (http://aws.amazon.com/1000genomes/), to create a more efficient way of managing, processing, and sharing data.
High-throughput sequencing will continue to transform biomedical sciences in almost every aspect [51] and provide new insights for our understanding of human diseases. For example, three groups recently reported discoveries of several new autism genes and suggested a much more complex disease mechanism, based on large-scale sequencing data of nearly 600 trios and 935 additional cases [52-54]. Along with the revolutionary discoveries based on NGS data, new tools and techniques will be developed to facilitate fast and accurate analysis.
Acknowledgments
We thank Dr. Gary A. Heiman and Alexandra Nguyen for providing valuable comments and proofreading the manuscript and the anonymous reviewers for their constructive advice. This work was supported by the National Institutes of Health (R00HG005846 to J.X.).