Storing Digital Information in Long-Read DNA
Article information

Abstract
There is urgent need for effective and cost-efficient data storage, as the worldwide requirement for data storage is rapidly growing. DNA has introduced a new tool for storing digital information. Recent studies have successfully stored digital information, such as text and gif animation. Previous studies tackled technical hurdles due to errors from DNA synthesis and sequencing. Studies also have focused on a strategy that makes use of 100‒150-bp read sizes in both synthesis and sequencing. In this paper, we a suggest novel data encoding/decoding scheme that makes use of long-read DNA (~1,000 bp). This enables accurate recovery of stored digital information with a smaller number of reads than the previous approach. Also, this approach reduces sequencing time.
Introduction
DNA is an excellent medium for archiving data [1]. Recent efforts have illustrated the potential for information storage in DNA. Martin Luther King’s ‘I Have a Dream’ speech and Shakespeare's sonnets have been recorded in DNA [2]. CRISPR-CAS technology has been introduced to store digital information, including digital movies [3]. Recent approaches enable polymerase chain reaction (PCR)‒based random access of stored information with 200 MB of digital data [4].
The principle of recent approaches is top synthesize specific sequences of DNA molecules and sequence them for writing and reading the digital data, respectively. However, both DNA synthesis and sequencing are highly error-prone. Various types of sequence alterations can be found, such as insertions, deletions, and substitutions, at rates of ∼0.01 errors/base [5]. Prior works have shown that a proper encoding scheme is necessary to recover the data under this noisy condition. Thus, existing approaches rely on a high degree of sequencing redundancy (i.e., having many copies of DNA molecules for each sequence and deep sequencing coverage).
Here, we present a new coding scheme for using long-read DNA sequences. Our approach explicitly reduces sequencing redundancy, requiring fewer resources and, in turn, fewer physical copies of any given DNA molecule to recover the stored data. In addition, using long-read DNA in conjunction with nano-pore sequencing technology shortens the sequencing time necessary to recover the information [6].
Our new coding scheme suggests a novel DNA fragment structure for long-read DNA data storage. The structure contains DNA sequences representing a data block, random linker, header, footer, and restriction sites. Because of the design, which contains restriction sites, each DNA fragment will serve as a unit that can be further ligated sequentially to form a longer DNA read. A longer DNA read can store a larger amount of digital information within a single molecule. To validate our scheme, we also provide practical assays to generate longer DNA molecules.
To investigate the challenges using long-read DNA for digital data storage, we encoded the first phrase of “Hun Min Jung Um” (118 characters). The entire information is stored in one long-read DNA sequence (1,872 bp). The read length is longer than previous approaches (150 bp). Our new encoding scheme reduces the sequencing depth required for data recovery (previous approach, 1,308; ours, 15) and sequencing time (previous approach, 3‒4 days; ours, days to 3‒4 h).
Methods
Storing binary information in a long-read DNA molecule
Binary data are encoded by two types of DNA fragments, each representing 0 or 1. Each fragment type will serve as a molecular unit to represent 0 or 1. A longer fragment that is composed of 0 or 1 DNA fragments can be synthesized and ligated to store more digital data in a single copy of a DNA molecule. Nanopore sequencing enables one to generate a long-read DNA sequence. Fig. 1 depicts a schematic diagram of the writing/recording strategy of this study.

Schematic diagram for writing/reading digital information in long-read DNA. (A) A unit DNA fragment storing digital information of ‘0’ or ‘1’ can synthesized and ligated. (B) DNA fragment storing digital information encoded by a series of unit DNA fragments can be read by nanopore sequencer.
DNA fragment unit for storage of binary information
In our scheme, 1 and 0 will be recorded in a specific sequence that we call the “signal,” flanked by random sequences that we call “noise,” in Fig. 2. The DNA sequence located in the signal part, “TATT” or “ACCC,” determines whether the DNA fragment represents 1 or 0. The noise region consists of a random linker that has a dissimilar sequence to the signal sequences providing space between signals. The noise region also contributes to DNA synthesis, reducing repeats.

DNA sequence design for storing digital information of 0 and 1. DNA fragment with a size of 16 bp will serve as a unit. To store digital information, units can be ligated or synthesized in the order of the digital information to store.
Digital information will be recorded in a DNA molecule that consists of restriction sites, a data block, and sequences to represent the start and end of the data block (Fig. 3). The data block contains a series of noise and signal units synthesized, as it depends on the digital data to store. DNA molecules can be further ligated to form a longer DNA molecule to store more digital data in a single copy of a DNA molecule.

Long-read DNA structure to store digital data. Short DNA fragments (size, 16 bp) that represent 0 or 1 will be located in a data block in a sequential order to represent digital information. ’Start’ and ’End’ are DNA sequences that mark the start and end of the data block, respectively. Flanking restriction sites are introduced for further ligation of long-read DNA to another.
Synthesis of DNA fragments
The length of the synthesized DNA is 780 to 880 bp, depending on the data block size. GC contents were adjusted to 50% to 54%, and plasmids were synthesized in pBHA. DNA synthesis was conducted by a company, Bioneer, that guaranteed delivery in 20 working days.
Ligation of DNA fragments in an ordered manner
Using a DNA fragment with a restriction site, a data block is to be further ligated in an ordered manner to form a longer DNA fragment. In our study, two DNA fragments with sizes of about 800‒1,000 bp will be cut with restriction enzymes to form a common ligation site. SpeI and XbarI are used for the first and second fragments, respectively. Fig. 4 describes this step, and the details of the restriction and ligation conditions follow.
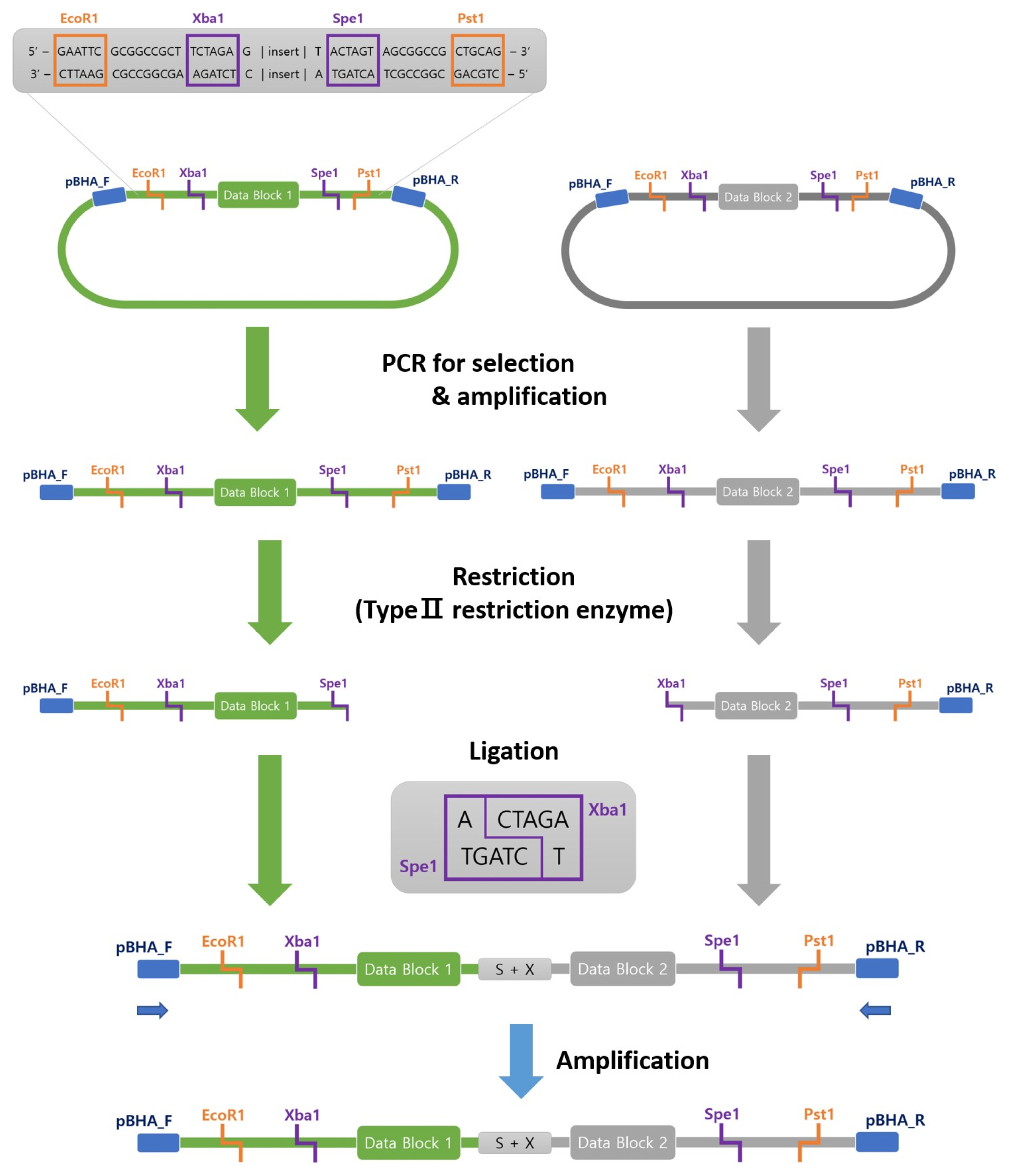
Long-read DNA can be further ligated to store digital information in a sequential order. PCR, polymerase chain reaction.
Amplifying DNA fragments using PCR
We designed a vector primer for the DNA fragment amplification. We used EmeraldAmp PCR Master Mix (cat. no. RR300A; Takara Bio, Tokyo, Japan), where the condition was one cycle at 95°C for 5 min; followed by 30 cycles of 30 s at 95°C, 30 s at 60°C, and 72°C for 1 min/kbp; and one cycle for 10 min at 72°C for definite extension. The PCR product was purified using an AccuPrep PCR/Gel DNA Purification Kit (cat. no. K-3037-CFG; Bioneer, Daejeon, Korea).
Restriction and ligation
3A assembly-associated enzymes were used: Xba1 (cat. no. R013S; Enzynomics, Daejeon, Korea) for the cut of the first restriction site and Spe1 (cat. no. R011S; Enzynomics) for the second. The cohesive DNA strand was ligated with AccuPower Ligation PreMix (cat. no. K-7103; Bioneer). The two DNA fragments were mixed at a 1:1 ratio and incubated ON at 4°C.
Extracting target DNA fragment
To separate between the ligated strand and non-ligated strand, we selectively amplified the ligation product using PCR. Electrophoresis and subsequent gel purification were conducted to yield the ligated strand.
Sequencing
Sequencing was performed using Oxford Nanopore NNGS equipment (Oxford Nanopore Technologies, Oxford, UK). First, we enriched the sample to meet the recommended concentration (1 μg of DNA or more). Second, we prepared a sequencing library with the LSK_108 kit from Oxford Nanopore. Third, we loaded the library into the single flow cell attached to the MinION equipment. We conducted the QC procedure to confirm if there were more than 8,000 active nanopores. We ran 2 h for sequencing, and about 10,000 reads were obtained for each sequencing run.
Filtering data and restoring binary information
We used two methods for restoring binary information. First, we used “Canu” software, which is a de novo assembler based on Linux [7]. The consensus sequence of de novo assembly is used to further detect “start,” “end,” 0, and 1 signals. We searched the signal sequence by allowing a 1-bp mismatch in the signal location.
Results
Long-read DNA preparation for digital data storage
To determine if our restriction and ligation conditions, we firstly optimized assay conditions with shorter fragments 490 bp, then further applied established conditions to form longer fragment with size of 1,895 bp. We confirmed the ligation product with gel electrophoresis, showing that both products were formed at the proper position of the molecular weight information (Fig. 5).

Confirmatory assay scheme for data storage. Two DNA molecules carrying digital information are ligated to form a longer DNA. (A) Expected size: start (35 bp) + end (35 bp) + information (54 bp) + random linker site (6 bp) = total 292 bp + 198 bp (TA cloning) = 490 bp. (B) Start signal (51 bp) + first DNA fragment (817 bp) + end signal (51 bp) + start signal (51 bp) + second DNA fragment (785 bp) + end (51 bp) + vector sequence (39 bp) = 1,845 bp.
Sanger sequencing
The short DNA fragment with a size of 490 bp was sequenced to confirm that our restriction and ligation assay did not distort the DNA sequence of our design scheme. The result showed 100% identity, which shows that the “start” and “end” random linker sequences and signal sequences for “0” and “1” were all well conserved after the assays for fragment preparation (Fig. 6).
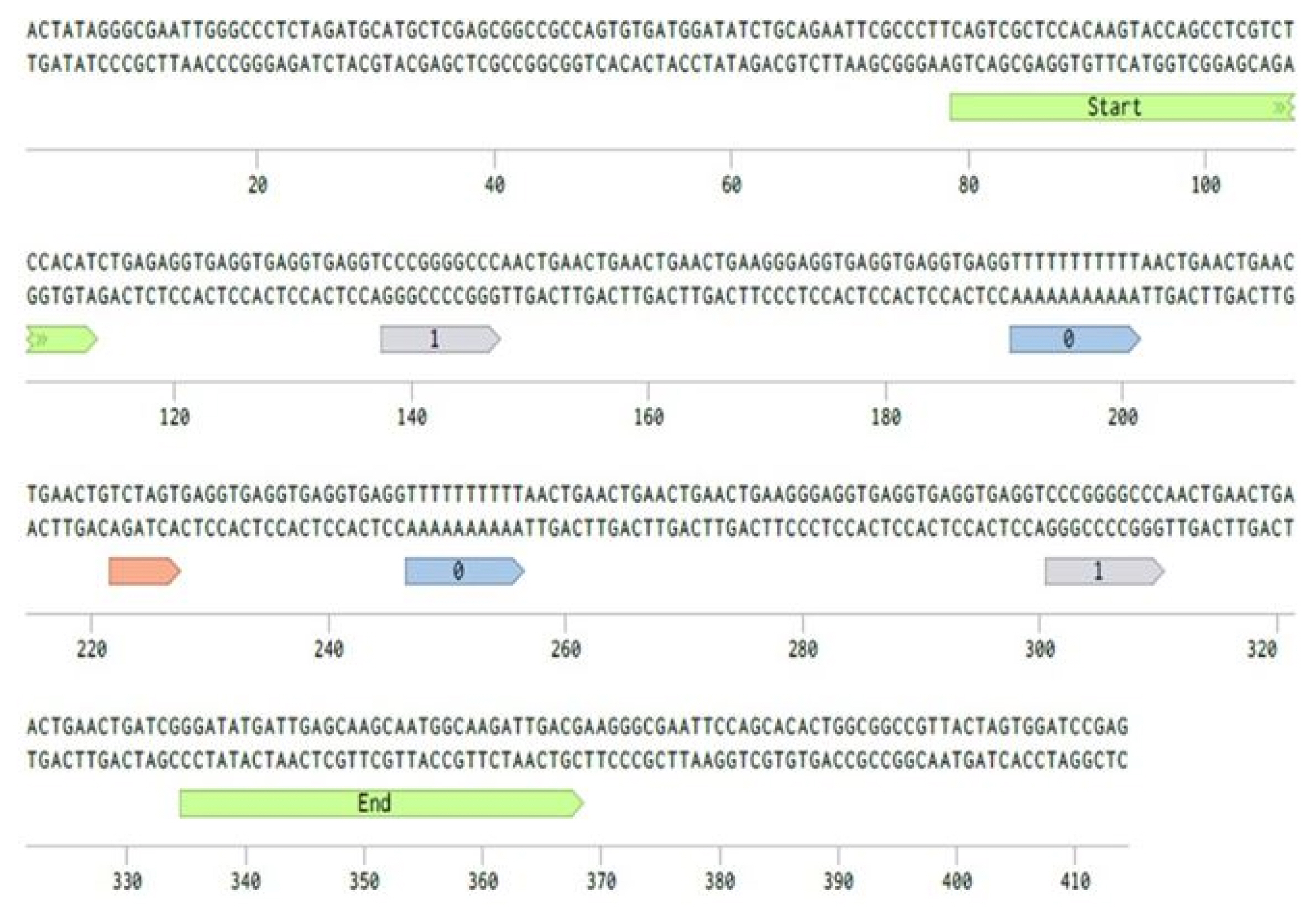
Validation of ligation assay by Sanger sequencing. Two DNA fragments carrying ’10’ and ’01’ information are ligated in an intended order. Our encoding scheme is marked with colors (green, start and end; blue, 0; gray, 1; and red, ligation site).
Nanopore sequencing
The DNA fragment storing digital information with a size of 1,854 bp, which is a longer length than the commercially available Sanger sequencing limit, was sequenced by Oxford nanopore sequencing method. The total number of reads obtained was 104,795. Out of 104,795 reads obtained, 675 reads were identical to the expected size of 1,854 bp. A total of 16,773 reads were found within 50 bp of the expected size (Fig. 7). There were also reads with lengths of 1,100 bp. Those reads were possibly from unligated product during the DNA fragment preparation process.
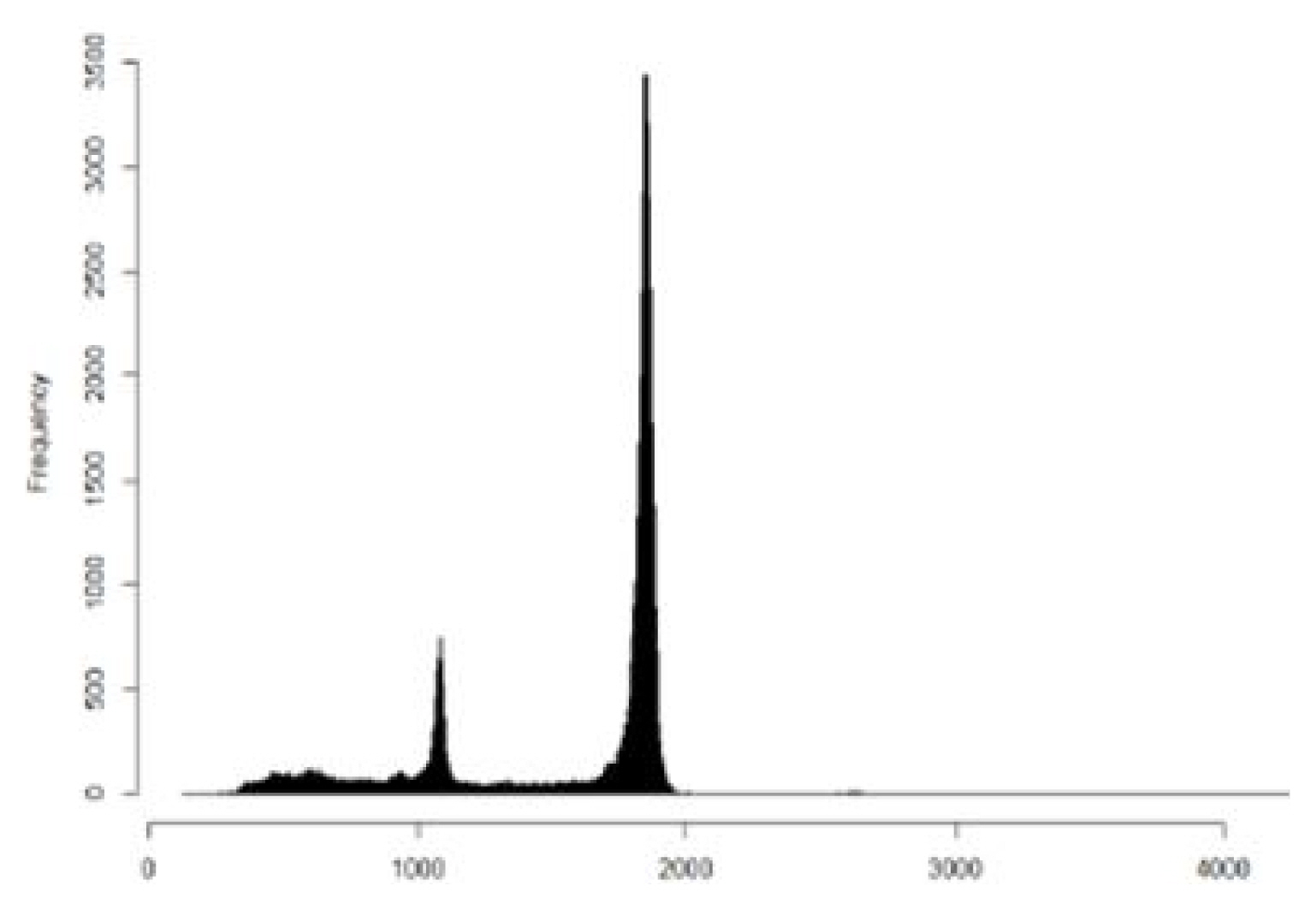
Read length distribution of nanopore sequencing. Most reads are found with a size of 1,854 bp.
Sampling of thousand reads their length is between 1,754 and 1,904 were assembled by “Canu” method to retrieve a consensus sequence. The consensus sequence showed 100% identity to the designed sequence that stored digital data. This means that digital information can be restored directly from the consensus sequence without any further sequence data treatment. However, sampling a fewer number of reads reduces the identity of the designed sequence.
We also considered 672 reads with the same length as the designed sequence. From these reads, we were able to successfully identify “start,” “end,” and “data block” sequences. The data block of the reads was aligned, minimizing Hamming distance. Voting of the aligned reads for a signal of “0” and “1” yielded the digital information intended to be stored. This shows that it is capable of reading digital information written in the long-read DNA by our method.
Our approach tolerates the low accuracy of nano-pore sequencing, which is capable of reading a long-read-length DNA up to 10 kb. Prior work on data storage in DNA molecules has been focused on using Illumina sequencing (Illumina, San Diego, CA, USA), which supports a shorter read length (∼150 bp) but provides high accuracy. Thus, our scheme is the first trial to enable writing DNA in longer fragments. Depending on the choice of sequencing technology, there are gains and losses. A comparison with prior work shows that our data storing scheme requires more base pairs to store 1 byte than other technology. However our method requires significantly less sequencing time to recover digital information (Table 1).

Comparison with prior work
Discussion
The need for data storage is rapidly and continuously growing. DNA data storage has the potential to eventually replace magnetic tape, the most commercially available storage for archiving.
We have proposed a new method for reading digital information into a DNA fragment. The method is different to prior work, which synthesized novel DNA sequences, depending on the digital data to be scored. Our approach is more scalable than prior methods, in that we can reuse pre-synthesized DNA for the data block (“0” and “1” signal sequences), which means there is no need to wait for de novo synthesis of DNA.
Using nano-pore sequencing with long reads has an advantage in the amount of times required to restore digital information from DNA molecules. Our current achievements show 3‒4 h for sequencing, which is much faster than prior works that made use of next-generation sequencing. As sequencing can be conducted in a parallel manner, our approach has more potential in restoring large amounts of data in a sort time.
We hope these advances may contribute to viable, large-scale DNA-based data storage.
Notes
Conflicts of Interest
No potential conflicts of interest relevant to this article was reported.
Authors’ contribution
Conceptualization: TJA
Formal analysis: TJA
Funding acquisition: TJA
Methodology: HB, HP
Writing – original draft: TJA
Writing – review & editing: HB, HP