Introduction
Fluoroquinolones (FQs) are one of the most extensively used antibiotics in human therapy and veterinary treatment [1]. These compounds are of exceptional interest because their clinical role has vastly expanded since the introduction of norfloxacin in the 1980s [2]. The antibacterial activity of FQs results from their inhibition of different kinds of enzymes, such as type II topoisomerase, DNA gyrase and DNA topoisomerase IV, which control DNA topology and are vital for chromosome replication and function [3]. Structural modifications to the key building block element of quinolone have led to an expanded antibacterial activity, optimal pharmacokinetics, and a better safety profile [4]. There are currently four generations of FQs and the first-generation drugs or the original quinolones were not fluorinated and included drugs like oxolinic acid and nalidixic acid. The next generation, which included fluorinated compounds exhibited enhanced antimicrobial activity against gram-negative bacteria and extended their activity to some gram-positive bacteria. These compounds are classified as second-generation FQs and include drugs such as ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin. An expanded activity against gram-positive bacteria, while retaining broad gram-negative coverage was exhibited by the third generation FQs, which included levofloxacin. Lastly, the fourth generation FQs, such as clinafloxacin, inhibited anaerobic bacteria, while retaining the gram-positive and gram-negative antibacterial activity [5, 6].
The four FQs in this study (ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin) belong to the class of second-generation FQs. Ciprofloxacin is an antibiotic that is widely used for the treatment of bacterial infection [7]. This includes skin infections, bone and joint infections and urinary tract infections, among others [8]. Ciprofloxacin is on the World Health Organization’s (WHO) “List of Essential Medicines” [9]. Lomefloxacin is a difluorinated quinolone used to treat bacterial infections including respiratory and urinary tract infections, and skin, eye, throat infections [10]. Ofloxacin is useful for treating bacterial infections of eye, ear [11], prostatitis [12], and certain types of infectious diarrhea [13]. Norfloxacin is used to treat urinary tract infections, gonorrhea and bladder and gynecological infections [14].
In this work, our interest is to study the antibacterial activity of FQs by molecular docking assays. We studied the inhibiting activity of the selected compounds with an antibacterial target protein. Furthermore, pharmacological studies of the compounds were performed. DNA gyrase has extensively been investigated as a target to study the mechanism of action for antibacterial compounds. FQs arrest the gyrase reaction after DNA cleavage leading to the generation of double-strand breaks [15]. To understand the interactions of the selected compounds at the active protein receptor sites (topoisomerase II DNA gyrase enzymes), docking studies were performed. Molecular docking provides a rational new approach to study the antibacterial properties of drugs [16]. A pharmacological analysis was also done for the second generation FQs [17]. This analysis included the evaluation of the pharmacological properties based on Lipinski’s rule of five, drug-likeness, bioactivity, and the drug score [18]. Furthermore, in silico preclinical trials including absorption, distribution, metabolism, excretion, and toxicity (ADMET) test, health effect probability, and maximum recommended daily dose (MRDD) were also predicted.
Methods
Antibacterial target protein retrieval and structural assessment
Topoisomerase II DNA gyrase is a well-known target protein for antibacterial studies, as also discussed in the introduction section above. The 3D structure of the topoisomerase II DNA gyrase enzyme (PDB ID: 2XCT) was downloaded from the Protein Data Bank [19] and imported and prepared by a multistep process using the protein preparation wizard of the WHATIF server [20]. The protocol was used to obtain the optimized and minimized energy conformation of the target protein. The DNA gyrase structure was extracted from its complex form. Following the extraction of the structure, it was subjected to energy minimization using the implementation of the optimized potential for liquid simulation (OPLS-2005) force field with implicit solvation. After that, the DNA gyrase structure was assessed by Ramachandran plot [21] and Verify3D [22].
Preparation of DNA gyrase protein structure for molecular docking
Prior to molecular docking, the topoisomerase II DNA gyrase protein was prepared to remove water molecules and unwanted ligands. All water molecules and small ions except the zinc ions were removed using the protein preparation wizard on the WHATIF web interface. Subsequently, hydrogen atoms were added to the structure to correct the tautomeric and oxidation states of amino acids residues.
Preparation of FQs compounds
The ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin compounds were prepared using the ChemDraw workspace and saved in a requisite format for the in silico assays. Ligands were used for the 2D to 3D conversion of compounds while the optimized potential for liquid simulation force field was used for conformation analysis. The ligands were prepared for molecular docking using the Swiss molecule preparation server.
Molecular docking studies
Molecular docking is an important computational technique in structural biology and computer-aided drug design. The major objective of molecular docking is to evaluate the feasible binding geometries of a putative FQ compounds with a target protein (known three-dimensional structure). To study the molecular interaction of FQ compounds with the topoisomerase II DNA gyrase enzymes, the crystal structure of topoisomerase II was downloaded from the protein data bank and molecular docking studies were performed using the HEX 8.0 program [23]. The HEX 8.0 CUDA program works by interacting complex simulations of steric shape conformation and the charge density of the structure. The binding residues and different bonds involved in the interaction of the resulting docked complexes were evaluated using the chimera molecular modeling package and the further docked complexes were visualized and analyzed using chimera [24].
Pharmacological analysis
The pharmacological properties and bioactivities of ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin were determined using various pharmacological methods. Pharmacological properties including the test based on Lipinski’s rule of five [25], bioactivity, drug likeliness, Ames toxicity, and the antibacterial properties were studied. Moreover, the ADMET test, the MRDD and other analyses were also studied using in silico assays with a variety of softwares such as SwissADME [26], ACD/I-Lab [27], Molinspiration, and FAF Drugs [28].
Results and Discussion
Antibacterial DNA gyrase of Staphylococcus aureus retrieval
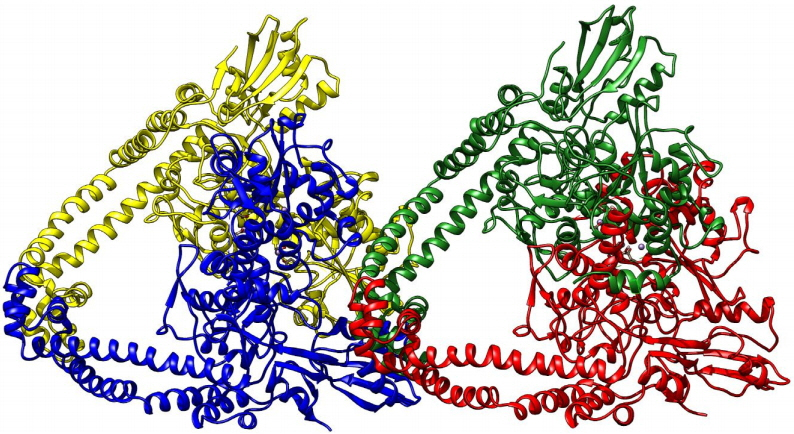
The three-dimensional structure of topoisomerase-II DNA gyrase enzyme of Staphylococcus aureus bacteria origin was retrieved from the protein data bank (PDB ID: 2XCT). The 3D crystal structure of this protein had a resolution of 3.35Å, and it is available in conjugation with small molecules, 1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-yl-1, 4-dihydroquinoline-3-carboxylic acid (CPF) and stabilized by manganese(II) ions. The topoisomerase-II DNA gyrase enzyme is composed of two subunits; one is the DNA gyrase subunit-A and another one is the DNA gyrase subunit-B. The 3D structure consists of four chains B, D, S, and U with a size of 692 amino acids (Fig. 1).
Topoisomerase-II DNA gyrase structural assessment
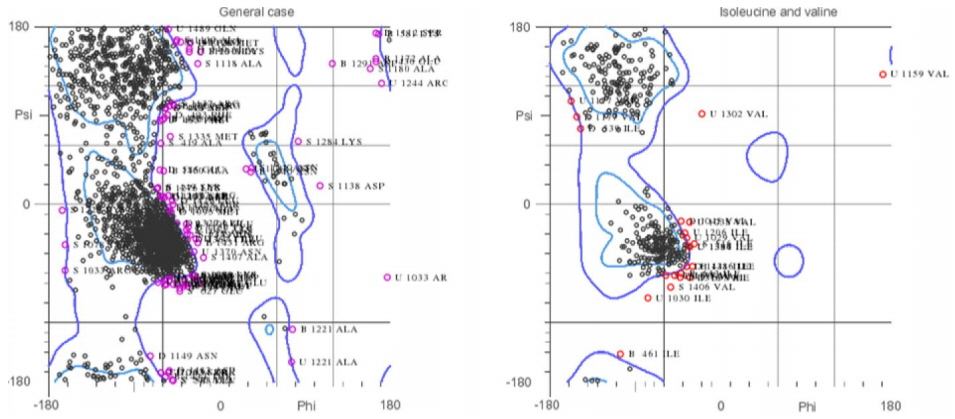
To carry out molecular interaction studies, the topoisomerase-II DNA gyrase enzyme of S. aureus structure was evaluated for its three-dimensional structural properties. The stereochemical properties of the DNA gyrase structure were analyzed via Ramachandran plot. The Ramachandran plots were used to assess dihedral angles and a number of amino acid residues in favored and disfavoured regions. The Ramachandran plot for topoisomerase-II DNA gyrase showed that most of the residues of the crystal structure are in the favored and allowed regions and a minimal percentage (4.8%) falls in the outlier region (Fig. 2). The Verify3D web interface confirmed the good quality of the DNA gyrase protein structure showing that more than 80% of the amino acids of the structure consist of 3D-1D profiles. Furthermore, X-ray diffraction studies showed that the crystal structure has a very low percentage (9.7%) of side chain outliers indicating that the DNA gyrase structure experimental values lie within the range of scores of the native protein structure of a similar size. The structural assessments carried out with the tools mentioned confirmed the good quality structure of the DNA gyrase enzyme and could be further used for molecular studies.
Binding studies of FQs compounds using molecular docking
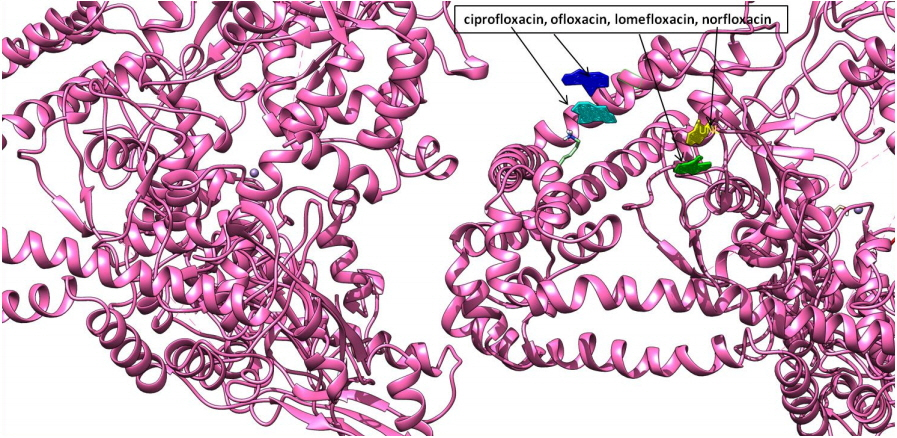
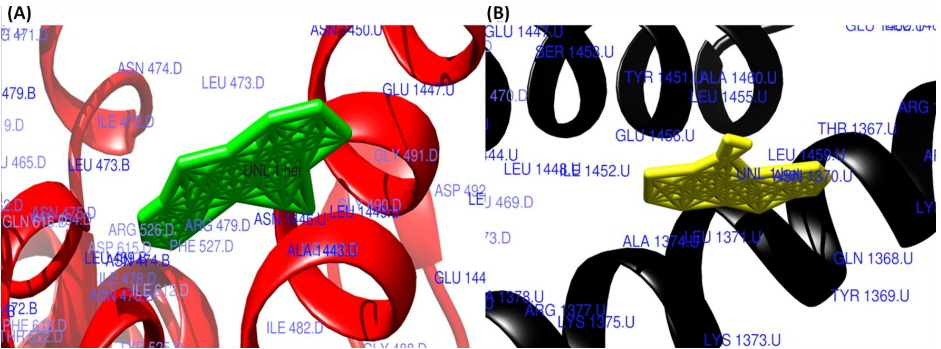
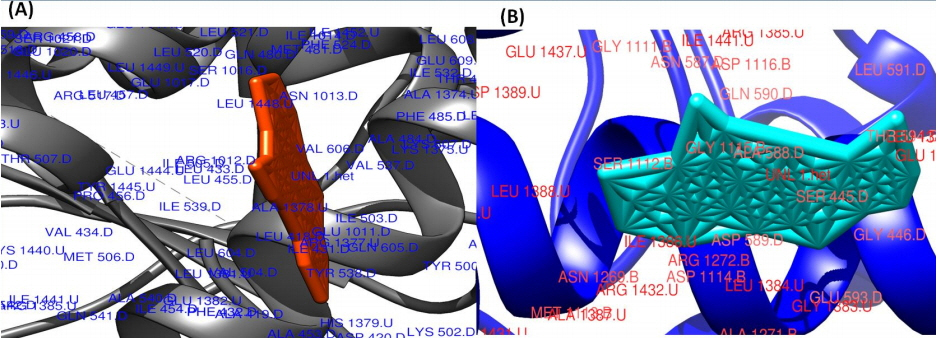
Molecular interaction studies of ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin compounds with the target antibacterial protein (DNA gyrase enzyme of S. aureus) were performed. The WHATIF server protein preparation wizard was employed to prepare the DNA gyrase enzyme. The prepared DNA gyrase structure was docked with ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin using the HEX 8.0 molecular docking software to obtain the best possible native conformation of the protein-ligand docked complex. The input parameters for the molecular docking were the receptor DNA gyrase structure PDB coordinate files and the ligand files for the ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin compounds with their set parameters. On the basis of the shape and electrostatics parameters, the total energy of interactions conformations was determined and the resulting final search was set to 25 (n = 25) conformations. The range of the molecular interaction angular search was confined by selecting the interface amino acid residues of the DNA gyrase enzyme structure with an angle range of 45°. The grid conformation box was placed on the interacting sites of the DNA gyrase enzyme structure, which provides space for compound rotation and translations. The other interaction parameters of interaction were set to the default values as given in the molecular docking suite. The molecular docking study showed that the ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin compounds have binding scores of −28.88, −38.52, −32.92, and −34.56 kJ/mol respectively. Among all four FQs compounds, the ofloxacin compound has the highest binding score (Fig. 3). The ofloxacin compound was found to interact strongly with the DNA gyrase structure residues at ASN 1013, VAL 606, VAL 537, ALA 1378, ILE 503, LEU 1448, LEU 418, and TYR 538 (Table 1, Figs. 4, 5). These molecular docking simulation studies suggest that the ofloxacin can be considered as a potential DNA gyrase enzyme inhibitor to be used as an antibacterial drug. Furthermore, these four compounds were evaluated for their pharmacological properties.
Pharmacological analysis of FQs compounds
Pharmacological analyses of the ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin compounds based on Lipinski’s rule of five were also performed. Lipinski’s rule of five evaluates the drug-likeness of a chemical compound and determines if a compound with certain pharmacological characteristics would be a possible orally active drug in humans. Drug molecules should be hydrophobic enough to last for the needed time in the body and not easily excreted, as this will enhance the bioactivity efficiency in the body. It is a good approach for drug lead design, especially for hydrophobicity and oral bioavailability prediction. Lipinski’s rule of five consists of the following predictions: molecular weight is from 160 to 500 Da, octanol-water partition coefficient (log P) is between −0.4 and +5.6, molar refractivity is from 40 to 130 cm3, hydrogen donor is less than 5 atoms, hydrogen acceptor is less than 10 atoms, and topological polar surface area is not more than 140 Å. The Molinspiration software was employed to study Lipinski’s rules for the ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin. It was found that all the four FQs compounds do not violate any part of Lipinski’s rule of five and also followed all the requisite properties needed for a potent drug. The results also indicate that compounds are hydrophobic as they have a low solubility in water. With their good hydrophobicity, it can be concluded that these compounds (ciprofloxacin, ofloxacin, lomefloxacin, norfloxacin) will be able to circulate long enough for the optimal duration and will be excreted conveniently. This will enhance their interaction duration with the target protein DNA gyrase enzyme and consequently, result in an efficient inhibition of the target antibacterial protein.
The other pharmacological parameters analyzed were the ADME properties of the FQ compounds. The adsorption properties of all four compounds were analyzed. These properties included the water solubility, Caco2 permeability, intestinal absorption in humans, skin permeability and P-glycoprotein inhibition.
All four compounds (ciprofloxacin, norfloxacin, ofloxacin, and lomefloxacin) were found to have high water solubility with values of −2.55, −3.03, −3.35, and −3.31, respectively. These values represent the high water solubility of FQ compounds at 25°C, defined on the basis of a database of more than 1,700 drug molecules.
The Caco2 permeability analysis showed that ofloxacin possesses a higher permeability (with a score of 1.402) than other FQs compounds studied. Caco2 permeability is an important property since these monolayers of cells are widely used in the study of the oral administration of compounds during the in vitro human model studies.
Another important consideration in the absorption of the drugs is intestinal absorption during oral administration. Ofloxacin was found to have a high absorption in the intestine with a high score of 96.964, as shown in Table 2. After that, the skin permeability of the compounds was studied, as it is an important parameter for the transdermal delivery of the drugs. The analysis revealed that ofloxacin has a good skin permeability in comparison to the other compounds, which possesses low permeability values. Moreover, all four compounds were found to possess the P-glycoprotein substrate characteristics with no inhibition of P-glycoprotein. These adsorption based pharmacological studies showed that all four compounds possess good adsorption properties, and ofloxacin possesses high-quality factors as a potential antibacterial drug.
After that, the pharmacological properties, including the distribution of drugs were studied, which include the volume of distribution (VDss), fraction unbound in human, blood-brain barrier (BBB) permeability, and central nervous system (CNS) permeability. The VDss reflects the drug volume needed so that the dose of the drug would distribute uniformly to provide a similar degree of concentration in the blood. Among the compounds, ciprofloxacin and ofloxacin showed good values for uniform distribution. The fraction of drug unbound in the human analysis showed that out of all the FQ compounds, ofloxacin has a minimal unbound fraction with a value of 0.375 (Table 3). This suggests that all four compounds have good distribution level pharmacological properties with very low BBB permeability and CNS permeability.
Next, the metabolism pharmacological parameters of four compounds were studied. All four compounds were not found to inhibit the cytochrome P450 and its different isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). This suggests that the noninhibition of cytochrome P450 will help in the metabolism of these compounds (Table 4). However, none of the compounds were found as a substrate of cytochrome P450. All four drugs (ciprofloxacin, norfloxacin, ofloxacin, and lomefloxacin) were also found to possess the optimal total clearance values 0.483, 0.361, 0.323, and 0.188 mL/min/kg respectively (Table 5). The total clearance reflects the hepatic clearance and renal clearance and indicates the requisite bioavailability property for uniform dosing concentrations. We found that all four compounds possess no violations of the Lipinski rule of five and other algorithms and high drug likelines with a large number of approved drugs.
Pharmacokinetic and toxicity analysis
The ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin compounds were further studied for their pharmacokinetic properties and toxicity using the pkCSM and FAF-Drug approaches. First, the Ames toxicity analysis of the compounds was performed, and it was seen that ofloxacin and norfloxacin do not show Ames toxicity. Since Ames test positive compounds may be mutagenic and may lead to cancer. Moreover, all four compounds were not found to inhibit the hERG-1 (human ether a go-go gene) and hERG-2. The inhibition of these regulatory potassium channels leads to the development of long QT syndrome to fatal ventricular arrhythmia.
Based on the hydrophobicity of the designed compounds, the oral bioavailability was also calculated using the FAF-Drugs and pkCSM software. The FAF-Drugs prediction showed that all four compounds have a good oral bioavailability based on Veber’s and Egan’s rules. Next, using the pkCSM software, the maximum recommended tolerated doses were calculated for ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin. All four compounds were found to have high oral rat acute toxicity values (2.311, 2.22, 2.237, and 2.216 log mg/kg/day, respectively). This indicates the high tolerance of compounds with recommended tolerated doses (Table 6). The results were based on the maximum recommended starting dose in the phase-1 clinical trials, based on the experimental data of 1,222 human clinical trials. Importantly, these compounds were also not found to cause any skin sensations. Lastly, we analyzed the antibacterial characteristics of the compounds against Tetrahymena pyriformis. We found that all the analyzed compounds (ciprofloxacin, ofloxacin, lomefloxacin and norfloxacin) have a high toxicity against the protozoa with a high IGC50 (concentration required to inhibit the 50% growth in log μg/L) score: 0.344, 0.292, 0.286, and 0.29 μg/L respectively. Moreover, the Minnow toxicity values for the compounds were calculated and showed that ciprofloxacin, ofloxacin, lomefloxacin, norfloxacin have values of 1.64, 1.3, 1.47, and 1.21, respectively. These results concluded that ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin have good bioactivity with significant pharmacological properties.
In conclusion, among the four tested FQs (ciprofloxacin, ofloxacin, lomefloxacin, and norfloxacin), ofloxacin exhibited a high antibacterial activity by in silico assays with high binding docking score of −38.52 kJ/mol to topoisomerase-II DNA gyrase (a well-known antibacterial target protein). All four analyzed FQ compounds meet Lipinski’s rule of five and show drug-likeliness. Based on our results, they also have a high passive absorption and good oral bioavailability based on Veber’s and Egan’s rules and is not toxic to humans.
The pharmacological and pharmacokinetic analysis of the compounds showed that all four compounds have good pharmacological properties. Moreover, ofloxacin was found to possess high IGC50 value of 0.286 μg/L against the T. pyriformis protozoa. Ofloxacin and norfloxacin compounds were tested to be negative for the Ames toxicity test, showing the non-carcinogenic character of the compounds. In particular, all four compounds exhibit marked activity against microorganisms, because they strongly interact with the active sites of topoisomerase II DNA gyrase enzymes and are supported by the optimal pharmacological properties.