Introduction
Since its advent, high throughput next-generation sequencing (NGS) technology has revolutionized the genomics research area, including transcriptome analysis and genome-wide association studies (GWASs) taking advantage of accelerated sequencing speed with reduced cost [1, 2]. Even though many bioinformatics software programs have been developed to handle and analyze the massive data generated from NGS, most of them are based on a command-line interface and require quite a high level of computational power [3], which creates a high barrier for wet lab biologists to enter into this field. Thanks to web-based analysis platforms, including Galaxy [4] and BIOEXPRESS [5], this barrier has been lowered. However, there still are problems. Downstream analyses, such as finding differentially expressed genes (DEGs), conducting Gene Ontology (GO) enrichment analysis, calculating linkage disequilibrium (LD), annotating gene information into GWAS results, and finally visualizing the resulting data, still require significant computer programming skills.
In this study, we present a dockerized application, IVAG. It provides a user-friendly web interface in which all downstream analyses mentioned above can be carried out without any programming knowledge. Detailed parameters for each analysis step can be adjusted with simple click-and-drag operation. IVAG interactively outputs publication-quality plots in response to the given parameters, and all of these plots can be downloaded. Also, a variety of data types, ranging from RNA sequencing (RNA-seq) and GWAS results to sequence read alignments, gene annotation, variant call information, and peak information, can be uploaded into the embedded genome browser and then visualized together to help users gain greater integrative insights into their data. Furthermore, IVAG is lightweight, allowing it to be deployed on a desktop computer, as well as a server application.
Methods
IVAG is mostly written in the R programming language [6] and dockerized [7] with all required dependencies to avoid compatibility issues. It uses the Shiny package [8] to build a user-friendly web interface and several other packages to analyze and visualize RNA-seq and GWAS data (Supplementary Table 1). VCFtools [9] and PLINK (v1.90b4.6) [10] were used for the LD analysis. The JBrowse platform [11] was integrated into IVAG, and all intermediate steps required for custom track construction were automated using a custom BASH script. Three publicly available plugins (Supplementary Table 2), with slight modification, were incorporated into the genome browser to build GWAS, GC content, and browser extensible data (BED) tracks. Gene transfer format (GTF)-to-general feature format 3 (GFF3) format conversion was carried out with Cufflinks (2.2.1) [12]. Binary sequence alignment map (BAM) and variant call format (VCF) files were sorted with SAMtools [13]. Example data were prepared using publicly available RNA-seq and GWAS data (Supplementary Table 3).
Results
Workflow
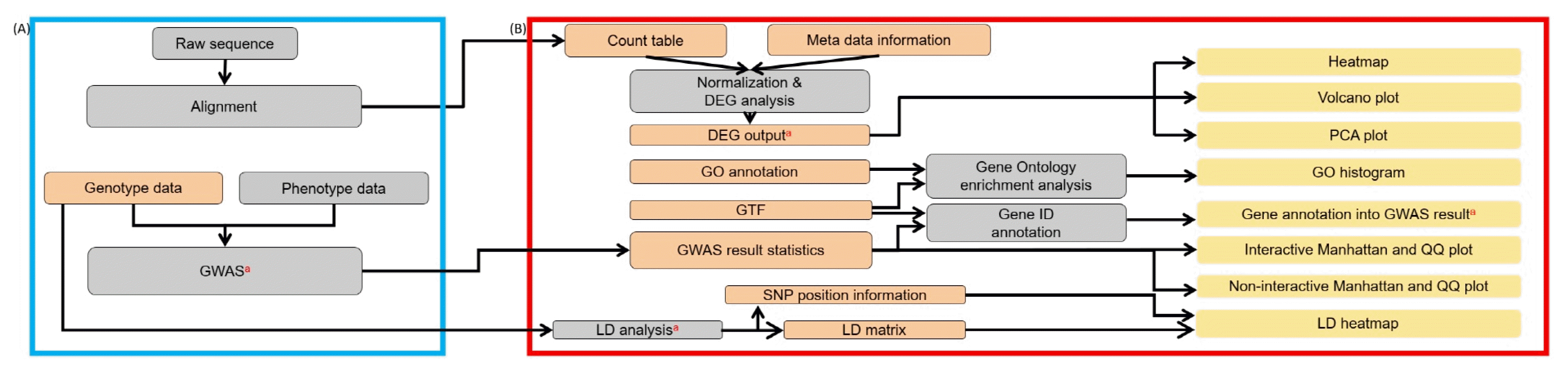
Fig. 1 shows a graphical overview of the pre-analysis steps and the IVAG workflow. The blue line (Fig. 1A) denotes a schematic representation of the external pipelines required for RNA-seq and GWAS data. These parts are prerequisites for downstream analyses prior to IVAG analysis. The red box in the right panel (Fig. 1B) illustrates the IVAG workflow. The orange items are input files for IVAG, and their detailed formats are described in Supplementary Figs. 1ŌĆō29.
RNA-seq
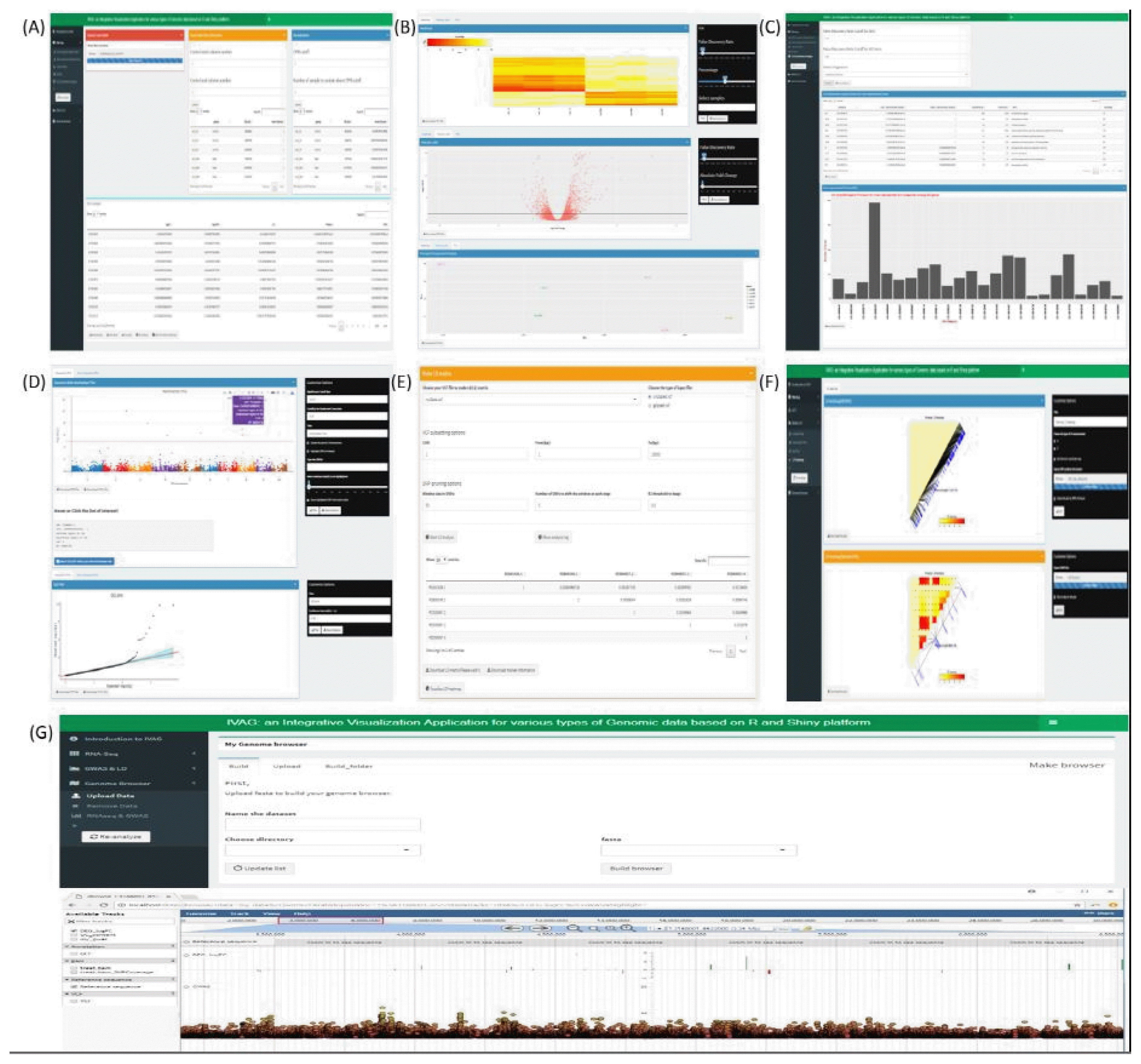
IVAG DEG analysis requires raw count RNA sequence data that can be generated using open source software, such as Htseq [14]. DEG analysis outputs a DEG results table generated with user-specified parameters based on the R Bioconductor package edgeR [15]. The output table consists of multiple columns, such as log2 fold-change, log2 count per million, associated p-value, and associated false discovery rate (Fig. 2A), and it can be visualized as a heatmap, a volcano plot, and a principal component analysis plot (Fig. 2B). The heatmap is generated using raw count data, which are converted to counts per million and normalized to a have row-based percentage value. The volcano plot is generated using log2 fold-change and the associated false discovery rate. Principal component analysis is generated using the raw count of each sample normalized to the log2 count per million. The heatmap and volcano plot can be interactively updated based on user-specified filtering criteria, such as false discovery rate or absolute fold-change. GO enrichment analysis uses the DEG analysis results table, GO annotation file, and GTF file. The DEG analysis results table can be generated using IVAG DEG analysis, or a pre-analyzed DEG analysis result can also be used. The GO annotation file consists of two columns: gene ID and GO category. A GTF file is needed to generate the gene length of each gene in the DEG analysis table. However, it can be omitted if a user wishes not to take gene length bias into account. IVAG GO enrichment analysis outputs over-represented and under-represented GO terms among DEGs (Fig. 2C) using the R Bioconductor package goseq [16]. It also shows a histogram of DEGs in each GO category based on its ontology: biological process, cellular component, and molecular function.
GWAS
Gene ID annotation requires a tab-separated GWAS result file comprising marker ID, chromosome ID, base position, and p-value columns in order, and a GTF file that contains strand and position information of genes. It returns a new GWAS result file in which gene, upstream, and downstream columns are added. Both GWAS result files, before and after this annotation, can be visualized in Manhattan and quantile-quantile plot with customizable options (Fig. 2D). One can see all information for a specific dot of oneŌĆÖs interest if he clicks on the interactive plots. The LD analysis part is read in a VCF file with several detailed options to generate an LD matrix and a marker information file, which can be visualized in the LD heatmap (Fig. 2E and 2F).
Genome browser
Constructing a custom genome browser with a reference genome sequence is the first step. After selecting one of the genome browsers configured in IVAG, various types of genomic data, including GTF, GFF3, BAM, BED, BigWig, and VCF, can be uploaded and visualized all together (Fig. 2G). Also, this genome browser receives RNA-seq and GWAS results generated from IVAG as inputs.
Discussion
IVAG is an easy-to-use, web-based application with three modules, including RNA-seq, GWAS, and a genome browser. This application enables scientists with little computational proficiency to analyze and visualize their data easily. Some web applications provide similar functions for RNA-seq and GWAS, but they have some limitations. For example, DEApp [17] provides differential expression analysis using three different methodsŌĆöedgeR, limma-voom, and DESeq2ŌĆöwhile a heatmap or a principal component analysis plot is not provided. START [18] can output several plots, but it does not offer a GO enrichment analysis function. LocusTrack [19] can visualize GWAS data and annotate multiple tracks on them, but it is limited to only one species, human. Zbrowse [20] can be used over every species. However, because it focuses on plotting multiple GWAS results in one panel to enable users to detect genotype-environment interactions, the number of markers that can be plotted for one trait is limited to 5,000. IVAG is not limited to a specific organism or the number of markers [14]. Most importantly, IVAG combines a genome browser with analysis and visualization modules so that users can analyze, visualize, and finally navigate their entire data interactively in one application. We offer only two analysis and visualization modules now, but several more modules are in development and will be included in the near future.