Introduction
It has been estimated that about 1.5% of human genomic DNA can be annotated as protein coding sequences [1]. So, more than 98% of the human genome does not encode protein [2, 3]. However, a large proportion of the genome transcribes non-coding RNAs such as miRNAs and long non-coding RNAs (lncRNAs) [4, 5]. LncRNAs have important roles in different cellular and molecular mechanisms [6]. These long RNAs regulate the activity and position of epigenetic machinery during cell function and segregation [7]. In fact, some of the lncRNAs can recruit catalytic activity of chromatin-modifying proteins [8]. Dysregulation of lncRNAs has been also reported in cancer initiation and progression. However, the molecular mechanism and regulation of these RNAs have been remained to be unknown [9, 10].
Rinn et al. [11] identified HOTAIR lncRNA with a 2.2 kb length. HOTAIR gene is located in a region between HOX11 and HOX12 on chromosome 12q13.3 [12–16]. HOTAIR lncRNA binds to both polycomb repressive complex 2 (PRC2) and lysine specific demethylase 1 (LSD1) complexes, through its 5′-3′ domains and directs them to HOXD gene cluster as well as other genes in order to increase gene silencing by coupling the histone H3K27 trimethylation and H3K4 demethylation [17, 18].
HOTAIR is an oncogene RNA that is known to have potential role in several cancers. Its overexpression is reported in different solid tumors such as breast, gastric, and colorectal tumors [19, 20]. The oncogenic role of HOTAIR is reported in different mechanisms such as cell proliferation, invasion, aggression, and metastasis of the tumor cells as well as inhibition of apoptosis [3, 21–25]. In spite of different reports on the potential oncogenic role of HOTAIR, the molecular regulation of this gene needs to be revealed by more studies.
Since the genetic and epigenetic complexities of the HOTAIR locus have not been characterized yet, we aimed to provide an integration data to highlight different compositional features of HOTAIR gene. The potential model may help to design future studies to reveal the molecular mechanisms of this lncRNA. In this study, we highlighted and described a number of features in HOTAIR locus, which may be involved in regulation of this gene. The integrated report is derived from the in silico approaches through different databases and software.
Methods
Different databases and bioinformatics software were used. Then, the data were reanalyzed and integrated in order to provide a potential model for describing the genetic and epigenetic features of the HOTAIR locus. Table 1 shows list of the in silico tools used in this study and the methodology is represented as a flowchart (Fig. 1). In our analyses, the desired sequence was mostly defined as a sequence that spans from 2 kb upstream of annotated transcription start site (TSS) of HOTAIR to the end of the gene. The selection was based on the previous studies defining putative promoter regions from −2 kb to +1 kb of the TSS [26]. Some data were analyzed through Encyclopedia of DNA Elements (ENCODE) project cited in University of California, Santa Cruz (UCSC) genome browser. Encode is a genome-wide consortium project with the aim of cataloging all functional elements in the human genome through related experimental conditions. In addition, all of the software was run with default parameters and criteria. The description of each software and database as well as their criteria of the analyses are described in below.
Results
HOTAIR gene is transcribed into different RNA isoforms by alternative compositional features
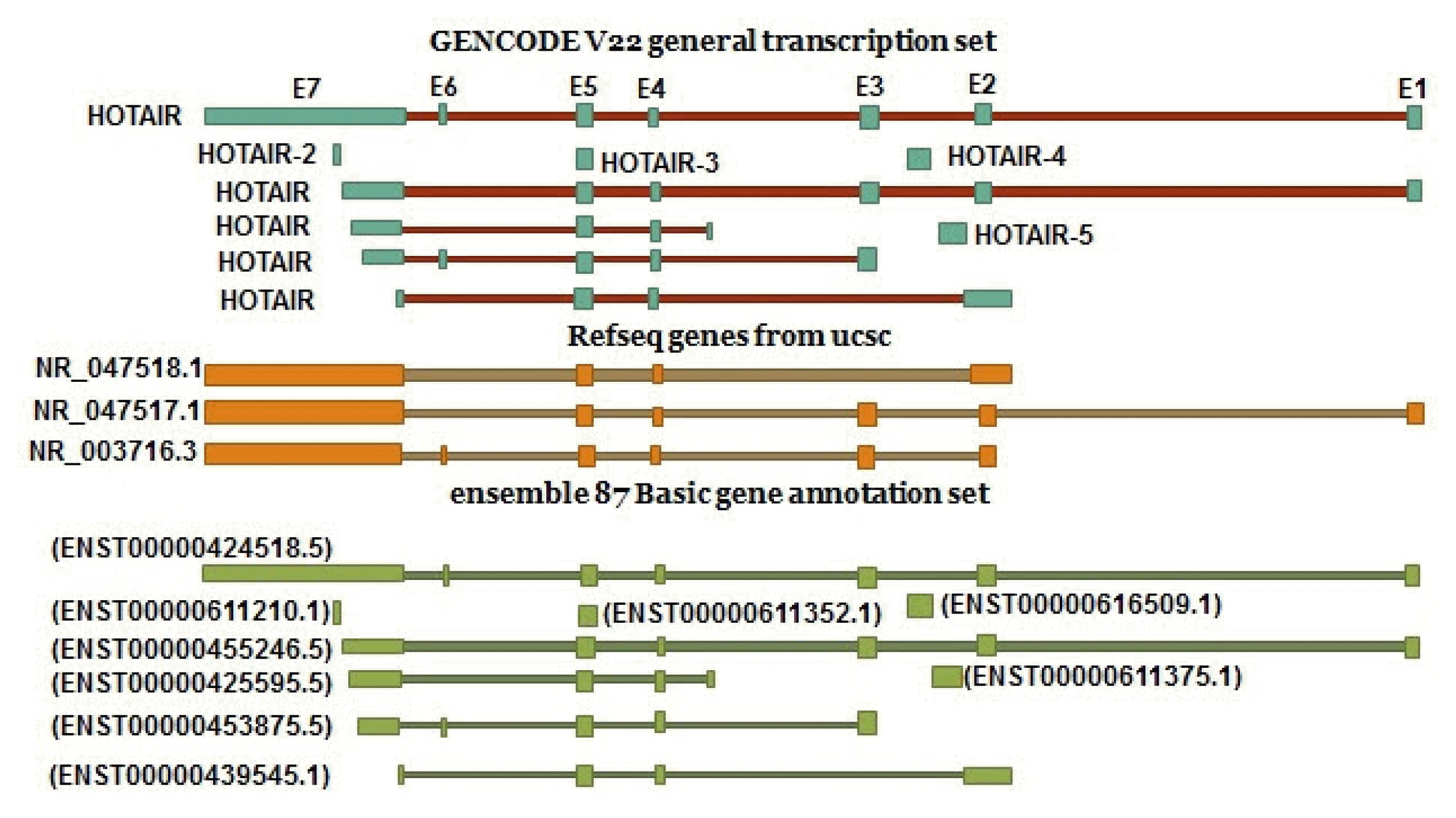
According to the Ace view database, 11 distinct GT-AG introns are identified in the HOTAIR gene. This results in seven different transcripts, six of which are created through alternative splicing (https://www.ncbi.nlm.nih.gov/ieb/research/acembly/). Different variants were found in GENECODE V22 and Ensembl. According to the Refseq, there are three transcript variants for this gene (NR_047518.1, NR_047517.1 and NR_003716.3) (Fig. 2).
Since it seems that alternative transcripts of HOTAIR are due to alternative promoters, TSSs, alternative polyadenylation sites, and alternative splicing, we tried to find different promoters, TSSs, polyadenylation, and splice sites in the HOTAIR gene.
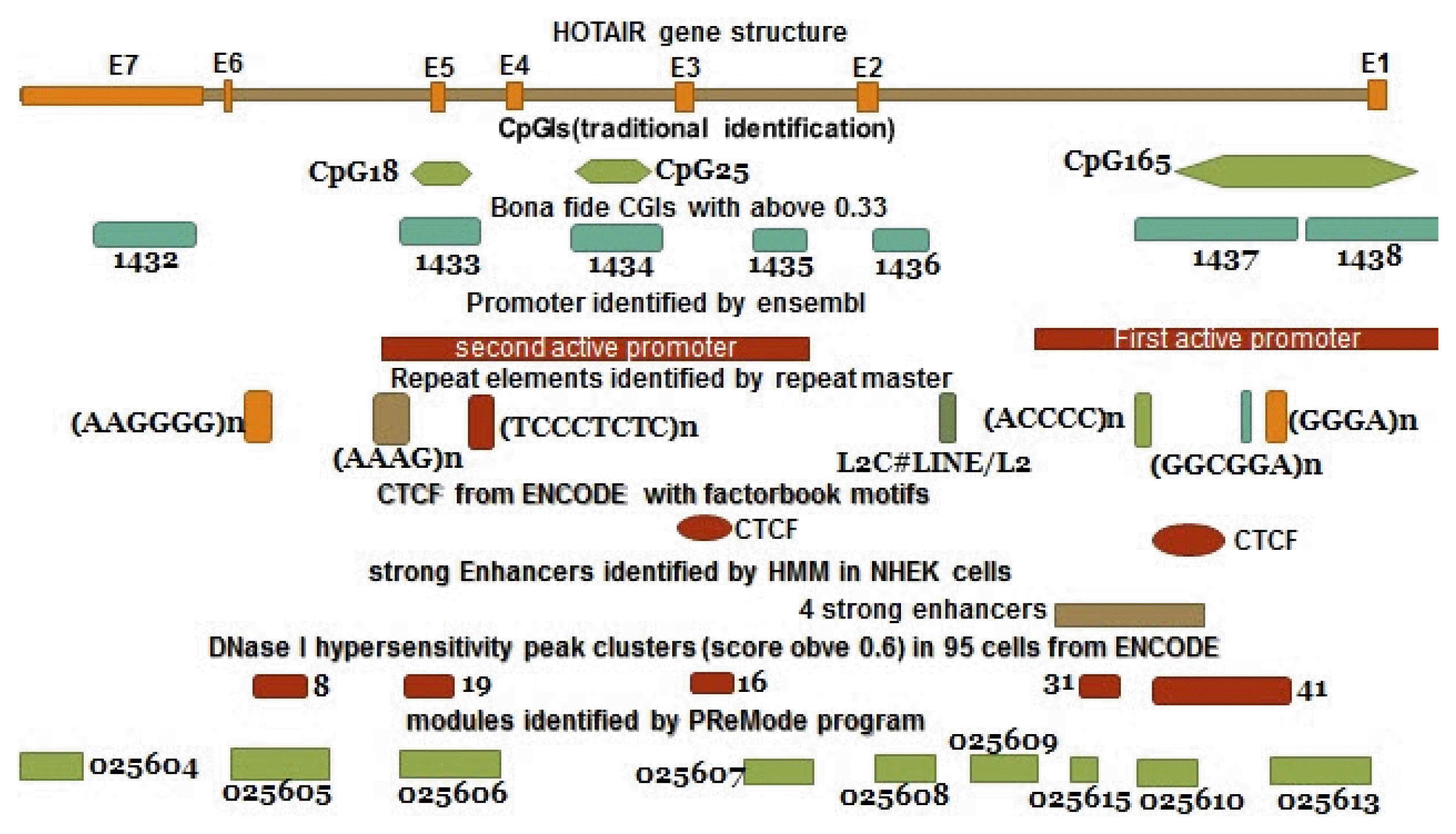
We found alternative promoters and polyadenylation sites in the HOTAIR locus (https://www.ncbi.nlm.nih.gov/ieb/research/acembly/). According to the Ensembl, there are two active promoters in this gene (Fig. 3). Also, Chromatin state segmentation using Hidden Markov Model (HMM) [27] identified these two active promoters as well as enhancers in the HOTAIR gene in some cell lines. The HMM is a probabilistic model representing probability distributions over sequences of observations. Supplementary Table 1 which is based on UCSC hg19, shows the positions of the active promoters of HOTAIR locus in Ensemble and HMM.
Promoter prediction with different tools recognized alternative promoters throughout this gene. Promoter scan program was run with the default promoter cutoff score. This program predicts promoters based on the degree of homologies with eukaryotic RNA pol II promoter sequences (https://www-bimas.cit.nih.gov/molbio/proscan/) [28]. Different TSSs were also found in the HOTAIR gene by different programs and software including Eponine, Switchgear, and Promoter 2 [29]. The Eponine program provides a probabilistic method for detecting TSSs. The Switchgear algorithm uses a scoring metric based largely on existing transcript evidence. Promoter2 takes advantage of a combination of principles that are common to neural networks and genetic algorithms. The positions of found TSSs compared to other features are shown in the Supplementary Table 1.
CpG islands were found to be overlapped with active promoters and DNase I hypersensitivity sites
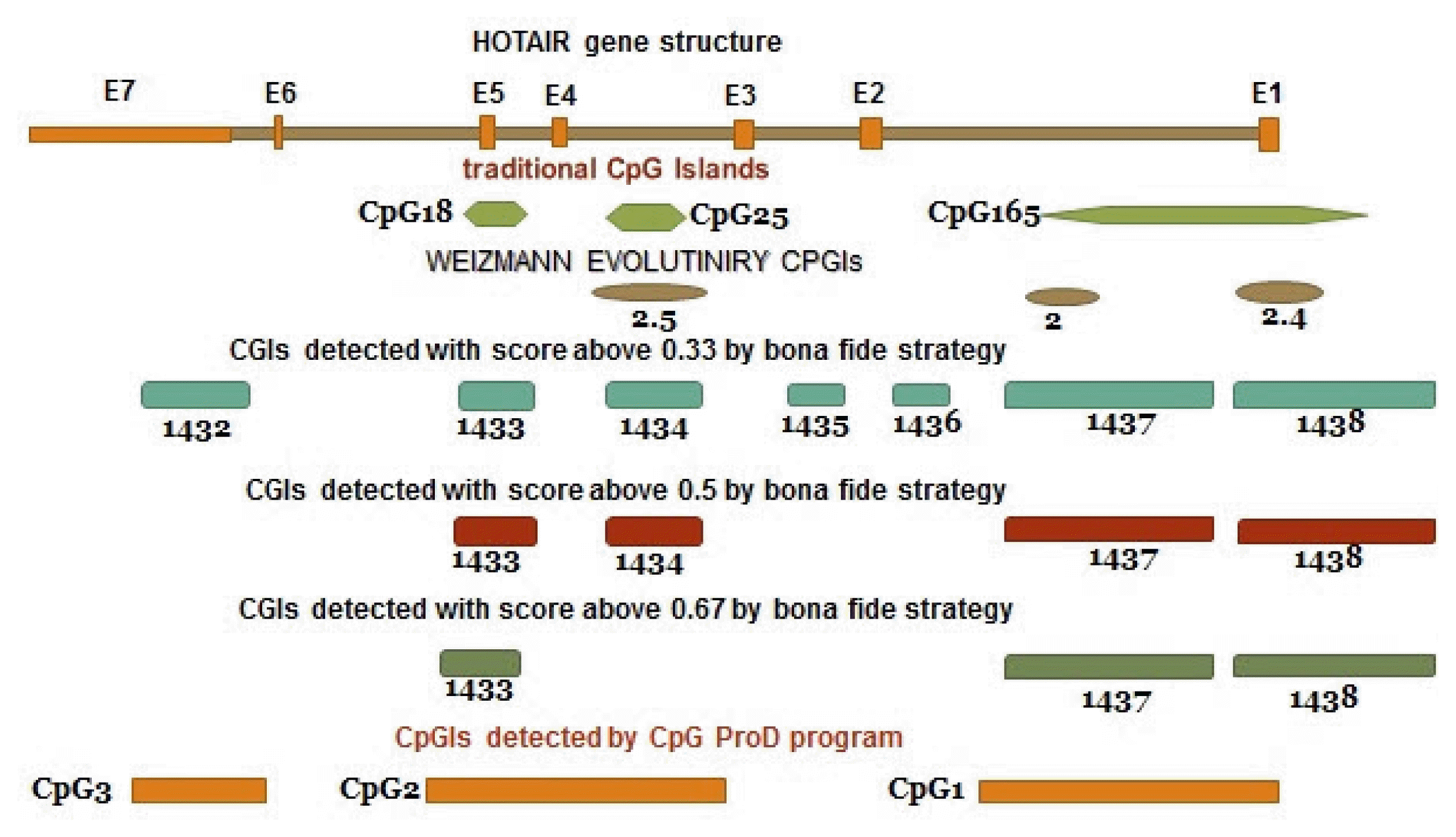
According to the UCSC browser, bona fide CpGIs, Weizmann Evolutionary, and CpG ProD program, there were different CpG Islands (CGIs) in the HOTAIR gene. These CpGIs are shown in the Fig. 4. UCSC genome browser identifies CGIs of human genome based on the regions of DNA with average (G+C) content greater than 50%, length greater than 200 bp and a moving average CpG O/E greater than 0.6 [30, 31]. “Bona fide” identifies functional CpGIs by linking genetic and epigenetic information [32]. Weizmann evolutionary (WE) predicts highly conserved CGIs through their classification of evolutionary dynamics (http://genome.ucsc.edu/) [33]. “CpG ProD” program identifies CpGIs-overlapping with promoters in the large genomic regions under analysis and shows these CpGIs with length longer than other CpGIs [34]. Then, we tried to find any overlap between CpGIs and other regulatory elements. Two TSSs (CHR-12-P0397-R1, CHR12-P0397-R2) were found within CpG165 (annotated in UCSC genome browser) and 1437 (derived from bona fide CGIs). The CpGIs were mostly overlapped with the active promoter regions (Fig. 3, Supplementary Table 1). We focused on CpG165 and found some regulatory elements which are within or near to this CpG (Table 2).
In addition, several DNase I hypersensitivity hotspots were found to be overlapped with CpGIs in some cell lines (Supplementary Table 1). We found the DNase I hypersensitivity peak clusters of HOTAIR gene in 95 cells with score greater than 0.6 by using UCSC genome browser. DNase I hypersensitivity peak cluster 19 is located within CpG1433 and mostly overlaps with CpG18. Also, DNase I hypersensitivity peak cluster 41 is located within CpG1437 and mostly overlaps with CpG165 and partially overlaps with CpG2 (WE) (Fig. 3, Supplementary Table 1).
Furthermore, we detected specific CpG dinucleotides methylation status within or near the predicted CpGIs in some cell lines by using ENCODE (Supplementary Table 2). This track identifies specific CpG dinucleotides methylation status by Infinium human methylation 450 bead array platform and classifies the methylation status into four groups: (1) not available (score = 0), (2) unmethylated (0 < score ≤ 200), (3) partially methylated (200 < score < 600), and (4) methylated (score ≥ 600) (http://genome.ucsc.edu/).
CTCF and transcription factor binding sites are overlapped with CpGIs and TSSs
GTEx RNA-seq strategy indicates that HOTAIR has variable expression in different tissues and its most expression level is in the artery-tibial tissue (data not shown). We found two putative regions for CTCF binding sites in the HOTAIR locus by ENCODE with factorbook motifs, one of which is located within CpG1437 (bona fide CpGIs) and mostly overlaps with CpG165 (Table 2, Fig. 3). This track determines regions of transcription factor binding sites taken from a comprehensive chip-seq experiments identified by ENCODE and factorbook pool (http://genome.ucsc.edu/). We predicted sequences of motifs and positions of these motifs in the HOTAIR locus by using MEME and MAST programs (Supplementary Table 3). MEME program searches the motifs from downloaded sequences through using complementary strengths of probabilistic and discrete models (http://MEME-Suite.org/) [35, 36]. The program was run with default parameters and normal mode of motif discovery. Mast program searches specific sequences based on predicted motifs by MEME program and exactly matches these sequences with the motifs sequences (http://MEME-Suite.org/) [37].
We found nine sequences of modules depending on their transcription factor binding sites in the HOTAIR locus by PReMode program [38, 39]. We observed some of these elements overlapped with the predicted CpGIs and TSSs (Fig. 3, Supplementary Table 1). In addition, we determined that some of these modules have common transcription factors (data not shown).
Some polymorphisms such as tandem repeats exist within the regulatory elements
Repeat Masker found several repeats sequences overlapped with regulatory elements of the HOTAIR locus such as CpGIs (Fig. 3, Supplementary Table 1) and motifs (Supplementary Table 3). Repeat master investigates query sequences and generates a detailed annotation of available repeats in these sequences and shows dispersed repeats and low complexity DNA sequences (http://genome.ucsc.edu/). In addition, tandem repeat finder, which analyzes simple tandem repeats, predicted one simple tandem repeat (GAGGGAGGGAGCGAGA) within this gene (Supplementary Table 1) (http://genome.ucsc.edu/) [40]. In addition, we found some simple nucleotide polymorphisms within regulatory sequences of HOTAIR gene (Supplementary Table 4).
Discussion
Studies have shown that aberrant epigenetic modifications including aberrant DNA methylation and histone modification are significantly involved in the dysregulation of genes with their potential roles in cancers [41]. However, identification of the exact elements of HOTAIR as well as their interaction has not been discovered yet. This study was aimed to find and highlight different regulatory elements by data integration. We identified putative regulatory elements that contribute to the regulation of HOTAIR expression by in silico analyses. Identification of these elements suggests new understanding of HOTAIR expression and might help to design future studies on this lncRNA which has oncogenic role in different cancers [42–45].
First, we tried to show different isoforms of HOTAIR RNA transcribed through alternative mechanisms. Since a recent study suggested the important role of HOTAIR domains in its function [46], we propose studying the molecular roles of different RNA isoforms in future researches. Then, in order to find alternative and potential features involved in generation of RNA isoforms, we checked the putative TSSs, promoters, and polyadenylation sites. We found different features, which are potentially involved in alternative transcription of HOTAIR gene.
Considering the potential involvement of methylation beyond CGI-promoters in human cancer, we focused on potential CGIs of HOTAIR. According to the fact that function of DNA methylation seems to be varied with context, we tried to find any relation between the CGIs and other compositional features such as TSSs, promoters, enhancers, DNase I hypersensitivity sites, and CTCF binding sites. Alterations in DNA methylation are known to cooperate with genetic elements and to be involved in human carcinogenesis. The results showed different CpGIs in the HOTAIR locus and determined their epigenetic status through integration analysis. The methylation status of these CGIs needs to be revealed in future researches. The methylation analysis will be so important because we currently know that most CGIs located in TSSs are not methylated. However, CGI methylation of the TSS is associated with long-term silencing. In addition, CGIs in gene bodies are sometimes methylated in a tissue-specific manner [47]. It has been reported that methylation of a CTCF-binding site may block the binding of CTCF. Altogether, different CpGIs overlapped with genetic elements seem to have important roles in controlling HOTAIR.
Some repeat sequences and single nucleotide polymorphisms exist within or next to the predicted CpGIs. We think that repeat number variations may effect on methylation status of regulatory regions of HOTAIR gene. Different studies reported some associations between polymorphisms of HOTAIR and cancers risks. The examples are the association between rs920778 [48], rs4759314 [49], and rs12826786 [25] and gastric cancer, rs7958904 and colorectal cancer [50], rs920788 and breast cancer [51], rs4759314 and rs7958904 in epithelial ovarian cancer [52]. We found that some SNPs are located within regulatory regions and so may effect on the gene expression. Also, since the repeat sequences of HOTAIR gene might contribute to the methylation status of regulatory regions, we highlighted the overlaps between these sequences and the predicted CpGIs.
Due to the overlap with active promoter, strong enhancer, CTCF binding site, DNase I hypersensitive sites, SNPs, and repeat sequences, CpG165 seems to be more important compared to other CpGIs for generation of the long RNA isoform. However, according to the Fig. 3, considering the overlap with other structural features, other CpGIs within the gene structure also seems to be involved in gene regulation. This integration model should be checked and validated in future experimental works.
Altogether, it seems that alternative transcripts of HOTAIR originate from interactions between genetic and epigenetic elements. Our data provide strong evidence based on the databases and in silico prediction that specific sequence motifs may potentially be involved in DNA methylation states of various set of CGIs in different tissues including normal and tumors. Our study suggests that the combinatorial binding of specific transcription factors plays a major role in regulation of HOTAIR expression. Future work that aims to provide detailed maps of epigenome in normal and diseased states is crucial to our understanding of HOTAIR role in cancer pathogenesis.