Introduction
Amino acids play a central role in the building block of protein. The primary structure of the protein is determined by the arrangement of 20 naturally occurring amino acids. The function of a protein is determined from their amino acids and also they depend upon interaction with cofactors, binding with metal ions and interaction with other proteins. The proteome of all the organism share significant metal ions and metal binding cofactors to carry out its essential function. It has been estimated that approximately 30% of all proteins contain at least one metal. The proteins play a vital role in biological processes and in the stability of the protein by binding with metal ions or metal containing-cofactors [1]. The proteins bind with major metal ions like transition metals, alkali, and alkaline metals. The frequent metal ions that bind with proteins are sodium, copper, iron, magnesium, manganese, potassium, and zinc ions respectively. In in-vitro condition, the unfolded polypeptide may are observed to interact with metal ions that direct the polypeptide folding process [2]. Identification of metal binding through experimental procedures like the use of metal ion affinity column chromatography [3, 4], electrophoretic mobility shift assay [5, 6], absorbance spectroscopy [7], gel electrophoresis [8], nuclear magnetic resonance spectroscopy [9ŌĆō11], and mass spectrometry [3, 12] require tedious steps and specific instruments, making them expensive and may be unsuitable for unknown targets. In this aspect, there is a need for computational predictors of protein binding metal ion in order to reduce time and cost. For example, predictions of protein metal binding ions are useful in structural genomics, to select proper growth medium for overexpression studies and for the easy interpretation of electron density maps. But fortunately, metal-binding ability are encoded in the amino acidic sequences and these primary sequences help in protein structure formation. Through genomic projects various organism genomic sequences have been annotated somehow along with metalloproteins contained in them [1]. Bioinformatics has been extensively used to predict metal-binding ability from amino acid sequences. Various computational methods like artificial neural networks [13], support vector machines [14], decision tree algorithm [15], graph theory [16], FoldX force field [17], CHED [18, 19], and geometry algorithm methods [16]. These methods depend upon either only sequence information or the use of both sequence and structure information. However, most of the available prediction methods are either based on the knowledge of the apoprotein structure or restricted to few specific cases, like the metal binding of histidines/cysteines. Most of these methods have been implemented as standalone software or web servers to the research community [15, 20].
Due to the availability of cheap and advancement of sequencing instruments, the sequence of proteins has increased rapidly over when compared to protein structure data. This due to the fact that experimental determining the three-dimensional of protein is difficult and expensive. Through various theoretical and experimental studies, it is proved that minimal set of the amino acid is sufficient for protein folding [21]. The minimal set of representative residues with similar features can be achieved by grouping together the 20 amino acids by clustering. This method is called as reduced or simplified amino acid alphabet. Several simplified amino acid alphabets have been proposed, which have been applied to pattern recognition method in the prediction of protein structure [22], for generation of consensus sequences from multiple alignments, and for protein folding prediction [23]. Various computational predictor has used simplified amino acids to predict the solubility on overexpression, remote homology detection [19], and identify the defensin peptide family [24], effects of cofactors on conformation transition [25], DNA-binding proteins [26], heat shock protein families [27], inter-residue interaction [28], protein adaptation to mutation [29], and protein disorder [30]. In the present study, a random forest algorithm has been deployed to predict metal ion binding protein based on the simplified amino acids proposed by Murphy et al. [21].
Methods
Dataset construction
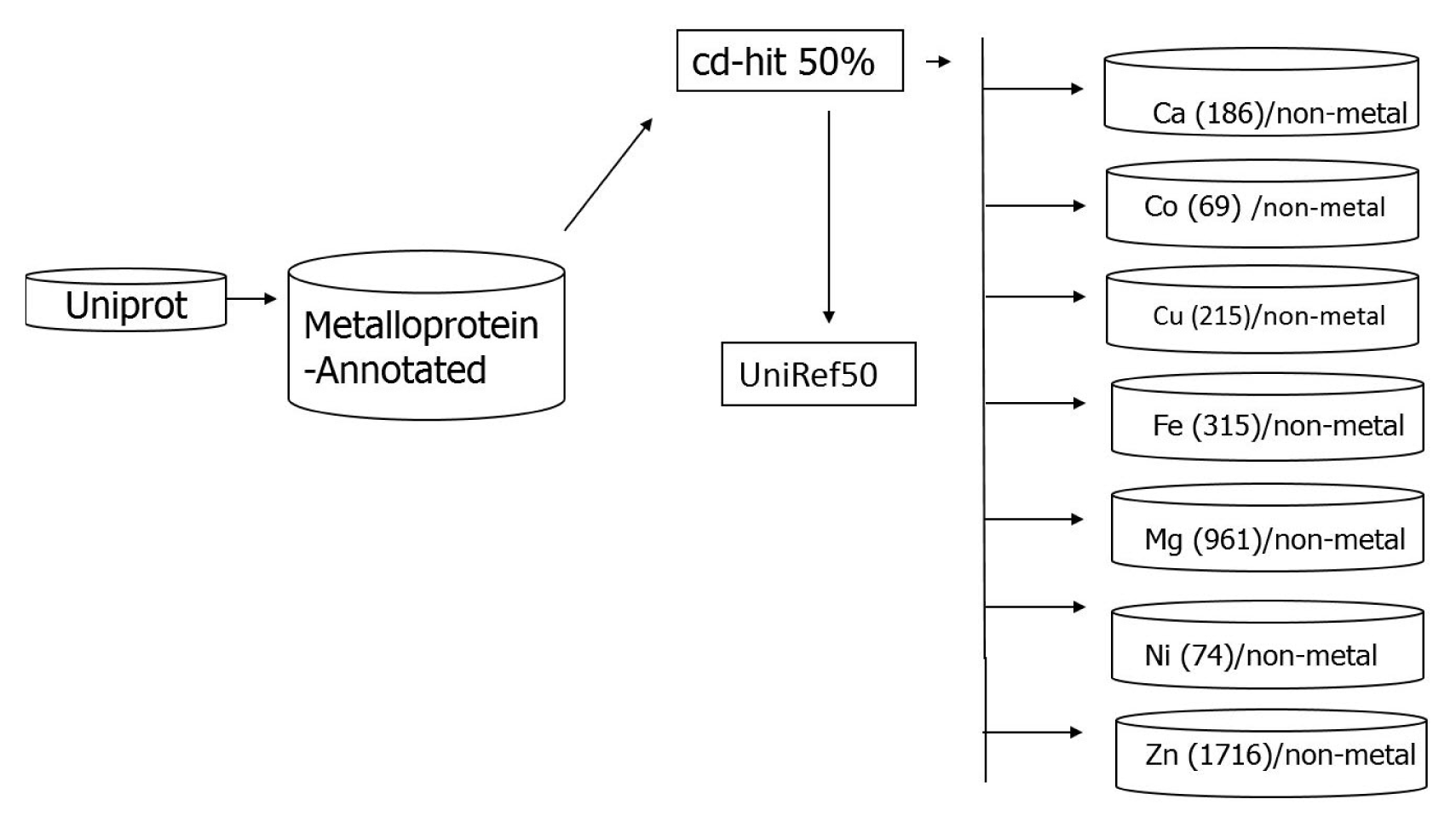
All the protein sequences were downloaded from the UniProt database [31] available at http://www.uniprot.org/. The downloaded sequences, annotated as metal containing, were grouped into eight subsets. Each of the subsets, containing one of the metal species viz., calcium, cobalt, copper, iron, magnesium, manganese, nickel, and zinc was considered to be metal-containing while all other entries were considered to be metal-free. Redundancy among the amino acid sequences was removed by clustering analysis using the cd-hit program [32] with the threshold of 50% level of percentage of identity, analogous by the UniRef 50 list [33] available in the UniProt database.
This resulted in eight data sets containing 186 calcium-containing proteins, 69 cobalt-containing proteins, 215 copper-containing proteins, 315 iron-containing proteins, 961 magnesium-containing proteins, 386 manganese-containing proteins, 74 nickel-containing proteins, and 1,716 zinc-containing proteins. All proteins containing calcium, cobalt, copper, magnesium, manganese, nickel, or zinc were then subtracted from the UniRef50 list, resulting in a collection of non-metalloproteins. The workflow of dataset construction is shown in Fig. 1. The problem of the imbalanced dataset can be solved as proposed by Cohen et al. [34]. Firstly, they pre-processes the data to re-establish class balance (either by upsizing the minority class or downsizing the majority class). Secondly, they modify the learning algorithm itself to copy with imbalanced data. In this study, we pre-processed the data which contains a balanced set of metal and non-metal ions. For this construction, nonŌĆōmetallo-proteins datasets sequences were randomly selected in order to have balanced set of metal and non-metal binding proteins for each metal ion, respectively.
Feature extraction by simplified amino acid alphabets
In order to investigate the effect of a particular class of amino acids on metal ion binding, the 20 amino acids were grouped into various classes based on certain common properties and the composition of the reduced sets of amino acids was considered. Feature extraction is done using the simplified amino acid alphabet. It estimates that reduced alphabets containing 10ŌĆō12 letters can be used to design foldable sequences for a large number of protein families. This estimate is based on the observation that there is little loss of the information necessary to pick out structural homologs in a clustered protein sequence database when a suitable reduction of the amino acid alphabet from 20 to 10 letters is made.
A simplified amino acid alphabet of 18 characters was used (Table 1). It is based on three independent amino acid classifications.
Conformational similarity
Conformational similarity indices are proposed by Chakrabarti and Pal [28] based on different residues are computed using the distribution of the main-chain and side-chain torsion angles and values have been used to cluster amino acids in proteins. In this method, the conformational similarity of the 20 amino acids based on torsion angles, which contains seven clusters ([CMQLEKRA], [P], [ND], [G], [HWFY], [S], and [TIV]) are used to represent variables.
BLOSUM 50 substitution matrix
The BLOSUM-50 matrix is proposed by Cannata et al. [35]. The matrix is deduced from amino acid pair frequencies in aligned blocks of a protein sequence database and is widely used for sequence alignment and comparison. The BLOSUM 50 matrix that they group together on the basis of the possibility of foldable structures and consists of the clusters: [P], [KR], [EDNQ], [ST], [AG], [H], [CILMV], and [YWF].
Hydrophobicity
The hydrophobicity scale by Rose et al. [36] is correlated to the average area of buried amino acids in globular proteins. This results in a scale which is not showing the helices of a protein, but rather the surface accessibility. It is based on the hydrophobicity scale which consists of the following cluster: [CFILMVW], [AG], [PH], [EDRK], and [NQSTY].
Random forest predictions
Random forest is a classification algorithm [37] that uses an ensemble of tree-structured classifiers. The random forest is a popular algorithm that has been used in designing computational predictors for various biological problems. Random forest is an ensemble learning method for classification. The random forest classifies a new object with an input vector, the input vector is predicted by each decision tree in the forest. Each tree provides a classification with votes and the class with most votes will be output as the predicted class. It is implemented by using Weka package [38, 39]. To ensure that parameter estimation and model generation of random forest is completely independent of the test data, a nested cross-validation procedure is performed. Nested cross-validation [40] means that there is an outer cross-validation loop for model assessment and an inner loop for model selection. In this study, the original samples are randomly divided into k = 10 parts in the outer loop. Each of these parts is chosen one by one for assessment, and the remaining nine of 10 samples are for model selection in the inner loop where a type of cross-validation using the so-called out-of-bag samples is performed.
Measurement of classifierŌĆÖs performance
When the predictor was focused on the problem of distinguishing proteins containing a certain type of metal ion from proteins that do not contain any type of metal, it is important that both sets contain the same number of proteins; otherwise, several figures of merit that are commonly used to monitor the prediction reliability would be seriously biased. The reliability of the predictions was monitored with the following quantities. If a protein of type 1 must be distinguished from a protein of type 2, a prediction was considered to be a true-positive if type 1 was correctly predicted; it was considered to be a true-negative if type 2 was correctly predicted; it was considered to be a false-negative if a type 1 protein was predicted to be a type 2 protein; and it was considered to be a false-positive if a type 2 protein was predicted to be a type 1 protein. Consequently, the following figures of merit, the sensitivity, the specificity, the accuracy, the Mathews correlation are computed [41] as shown in the Eq. (1) below.
Results and Discussion
By using a simplified amino acid alphabet based on three independent amino acid classifications, amino acid cluster variables were obtained. Conformational similarity contains seven clusters: [CMQLEKRA], [P], [ND], [G], [HWFY], [S], and [TIV]. BLOSUM 50 substitution matrix contain [P], [KR], [EDNQ], [ST], [AG], [H], [CILMV], and [YWF]. The hydrophobicity scale contains [CFILMVW], [AG], [PH], [EDRK], and [NQSTY]. Out of 20 amino acid clusters, cluster [P] and [AG] which are present in more than one simplified alphabet were considered only once and these results in 18 variables (Table 1). The 18 variables are represented with percentage of occurrence as follows.
The percentage of occurrence pcaa,i of the amino acid aa in the ith protein was computed for each of the 20 types of amino acids in each protein as per Eq. (2). The protein sequences represented by the amino acid percentage of occurrence using 18 variables were employed with random forest algorithm using Weka suite. The metallo-proteins were identified using all the 18 variables with high accuracy ranging from 69% for zinc and 90% for nickel (Table 2). Moreover, prediction performance was studied by feature selection method by removing one variable at a time and maintaining the highest value in performance indices. Measurements are removed until there is an unacceptable degradation in system performance. The use of feature selection method will eliminate alphabets which are irrelevant or redundant features, and thus it improves the accuracy of the learning algorithm. To select an optimal subset of variables, we first analyzed how individual attributes from the initial set of 18 variables, contributed to predictive accuracy. For feature selection, we employed the wrapper approach as it uses the learning algorithm to test all existing feature subsets. The wrapper method will use a subset of features to train the model. Based on the inferences, the feature can be added or removed to improve the accuracy of the learning algorithm. We used a backward feature elimination, by starting with the full set and deleting attributes one at a time for searching the feature space [42, 43].
The specific steps of the wrapper approach followed in this study.
Partitioning the data with 10-fold cross-validation (k = 10).
On each cross-validation training set, the learning machine was trained by using all 18 variables, to produce a ranking of the variables according to the importance. The cross-validation test set predictions were recorded.
Then the variables are removed which are least important one by one and another learning machine was trained based on remaining variables, the cross-validation test set predictions were once again recorded. This step is repeated by removing each variable until at small number remain.
Aggregate the predictions from all 10 cross-validation test sets and compute the aggregate accuracy at each step down in a number of variables.
By the following the above steps, feature selection of variables was done by wrapper approach employing random forest machine learning algorithm. Based on aggregate accuracy, the important variables for copper ion prediction are PH variable and least preferred variables are AG and CMQLEKRA (Table 3). Based on Table 3, it is understood that removing PH variable decrease the accuracy of the classifier whereas removing AG and CMQLEKRA improves the accuracy of the classifier. For calcium ion prediction, the least important variable is P and EDNQ; removing these variable improves the performance of the classifier (Table 4). Similarly, for cobalt ion prediction, the variable CILMV is the least preferred variable as it affects the performance of the classifier (Table 5). For iron ion prediction, removing variable CFILMVW improves the performance of the classifier (Table 6). For magnesium, ion prediction variable ST and ND are least preferred variables (Table 7). For manganese ion prediction, removing variable FWY improves the accuracy of the classifier (Table 8). For nickel ion prediction, variable EDRK is the least preferred one (Table 9). For zinc ion prediction, the least preferred variable is HWFY (Table 10).
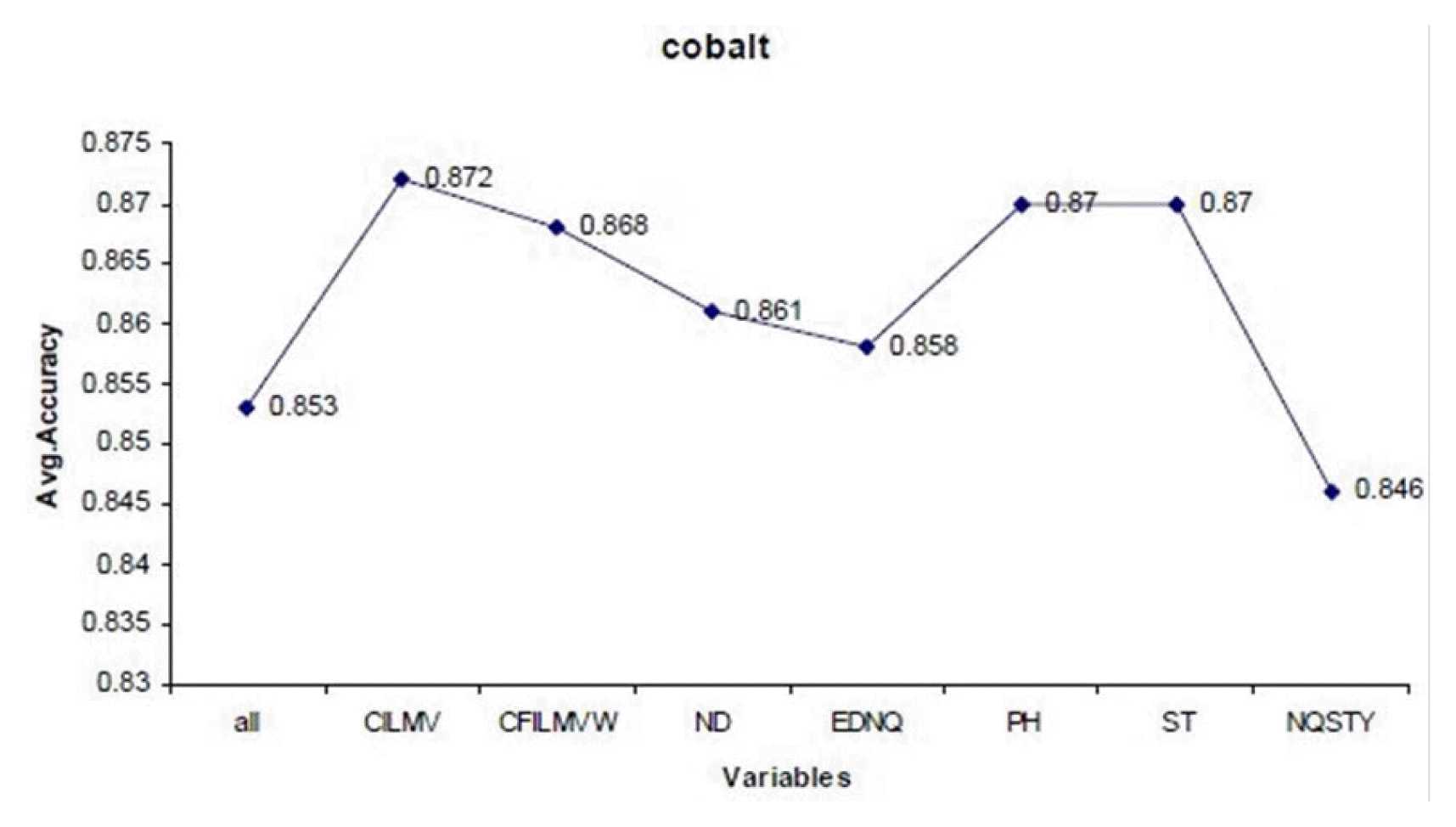
For example, cobalt metal binding protein can be discriminated from non-metal ions with all 18 variables with the accuracy of 85% (Fig. 2). It can be seen that, on removing variable V14 (CILMV) from the subset, the accuracy of the predictor improves from 85% to 87%. After removing of variables V8 (CFILMVW), V3 (ND), V17 (EDNQ), V10 (PH), and V16 (ST), the accuracy values are in the range from 86% to 87%. There is a drastic decrease in accuracy of the classifier by removing the variable V12 (NQSTY) to 84%. No further reduction of the set was possible, as the performance of random forest classifier dropped if any further attributes were eliminated. It can be seen that accuracy of prediction of metal binding proteins can be improved (e.g., calcium from 74% to 77%, cobalt from 83% to 85%, and nickel from 69% to 77%) by elimination of certain noisy features, up to certain limit and further improvement is then impossible. According to this backward strategy of feature selection, it can be observed that the prediction performance can be slightly improved. Some common variables rejected are V14 (CILMV) in calcium and cobalt, V8 (CFILMVW) in copper and iron.
In this work, a new random forest based approach is developed combining hybrid feature of simplified amino acid alphabets for prediction of metal ion binding sites of iron, copper manganese, magnesium, nickel, calcium, cobalt, and zinc from amino acid sequence data. The result indicates that the random forest model has a high prediction accuracy in predicting metal ion binding sites. These metal binding prediction methods are helpful to avoid the selection of ŌĆśimpossibleŌĆÖ targets in structural biology and proteomics.