Introduction
DNA damage occurs naturally and due to the environment, altering the cell’s abilities that are encoded by the DNA, and may lead to diseases, like cancer. Cells respond to DNA damage by DNA repair and cellular apoptosis [1, 2]. Apurinic/apyrimidinic endonuclease (APE) is an enzyme that identifies damaged apurinic/apyrimidinic sites in DNA, cuts the phosphodiester bond in the backbone of the sites, and has critical roles in the base excision pathway [3]. APE1 has recently been noted as a potential drug target for treating cancer, in that overexpression of the enzyme has been observed and shown to be associated with a poor response to cancer treatment, such as radiation and anticancer drugs, and a lower overall survival rate [4–7]. Antineoplastic agents that are to treat cancers are known to induce the expression of APE1, increasing the resistance of tumor cells to drug treatment. Thus, compounds that inhibit the activity of APE1 could be potential anticancer drugs with DNA-damaging antineoplastic agents used in the clinic [8].
For this reason, there have been several attempts to develop compounds targeting APE1. Currently, although there is no approved drug yet, three candidates—7-nitroindole-2-carboxylic acid (also known as CRT0044876) [9, 10], lucanthone (also known as Miracil D) [9], and methoxyamine (trademark TRC102)—are known to inhibit APE1 activity and are under examination in clinical trials. Lucanthone and CRT0044876 have rings similar to the deoxyribose sugar ring without a base and many hydrogen bond acceptors that can interact with hydrogen bond donors in the active site of APE1. These properties lead APE1 to stick in the site and prevent it from repairing DNA damage [11]. Methoxyamine is known to attack the open-ring form of AP sites to form an oxime linkage. In other words, methoxyamine blocks APE1 from accessing the lesion site rather than targeting the enzyme directly. This may lead to nonspecific off-target effects [12, 13]. Although several inhibitors of APE1 have been discovered, most potent compounds have weaknesses [14]. Thus, it is necessary to find novel kinds of potential inhibitors targeting APE1. Here, we present out work, in which we applied pharmacophore modeling and virtual screening. The overall procedures we carried out are illustrated in Fig. 1. We constructed pharmacophore models by capturing the common features of known inhibitors of APE1. The modes were used to screen a vast number of lead-like compounds, and molecular docking was used to prioritize the hits of the screen.
Methods
Selection of ligands for pharmacophore modeling
From the ChEMBL [15] database, we retrieved 52 compounds known to be targets of APE1 and 51 compounds with an IC50 of less than 10 μM. By eliminating redundancy, the number of compounds was reduced into 83. The list did not contain methoxyamine; so, methoxyamine was also added to the list.
We clustered these 84 compounds by Tanimoto distance, based on the PubChem fingerprint [16], and finally categorized them into two groups by excluding two outliers (CHEMBL1213633 and CHEMBL313493) (Fig. 2). A total of 49 molecules in group 1 (Fig. 3) and 33 molecules in group 2 (Fig. 4) were used to generate pharmacophore models 1 and 2, respectively.
Generation of pharmacophore model
Ligandscout tools 4.1 [17] was used to generate the ligand-based pharmacophore models. Ligandscout is known to be able to increase the selectivity of a pharmacophore model with the excluded volume feature. To generate more flexible pharmacophore, the threshold of the portion of partially matching features was set to 20%.
Pharmacophore screen
For the initial set of the pharmacophore screen, we selected a lead-like subset [18], defined by the ZINC database (ZINC is not commercial) [19]. Similar to druglikeness [20], like Lipinski’s rule of 5 [21], lead-like compounds are defined as being large enough to be validated in experiments but are smaller than most drugs, optimized too specifically, and more soluble than their drug-like compounds. ZINC provides a lead-like subset fulfilling leadlikeness as follows: (1) molecular weight between 250 and 350 Da, (2) partition coefficient log p ≤ 3.5, and (3) no more than seven rotatable bonds. The structures of lead-like compounds in medium pH were downloaded and converted into a database for screening by idbgen, a component of Ligandscout. We carried out pharmacophore screens using iscreen in Ligandscout for models 1 and 2 independently. Pharmacophore fit scores were also calculated by LigandScout based on the number of matching pharmacophore features and the root-mean-square deviation of the pharmacophore alignment.
Results and Discussion
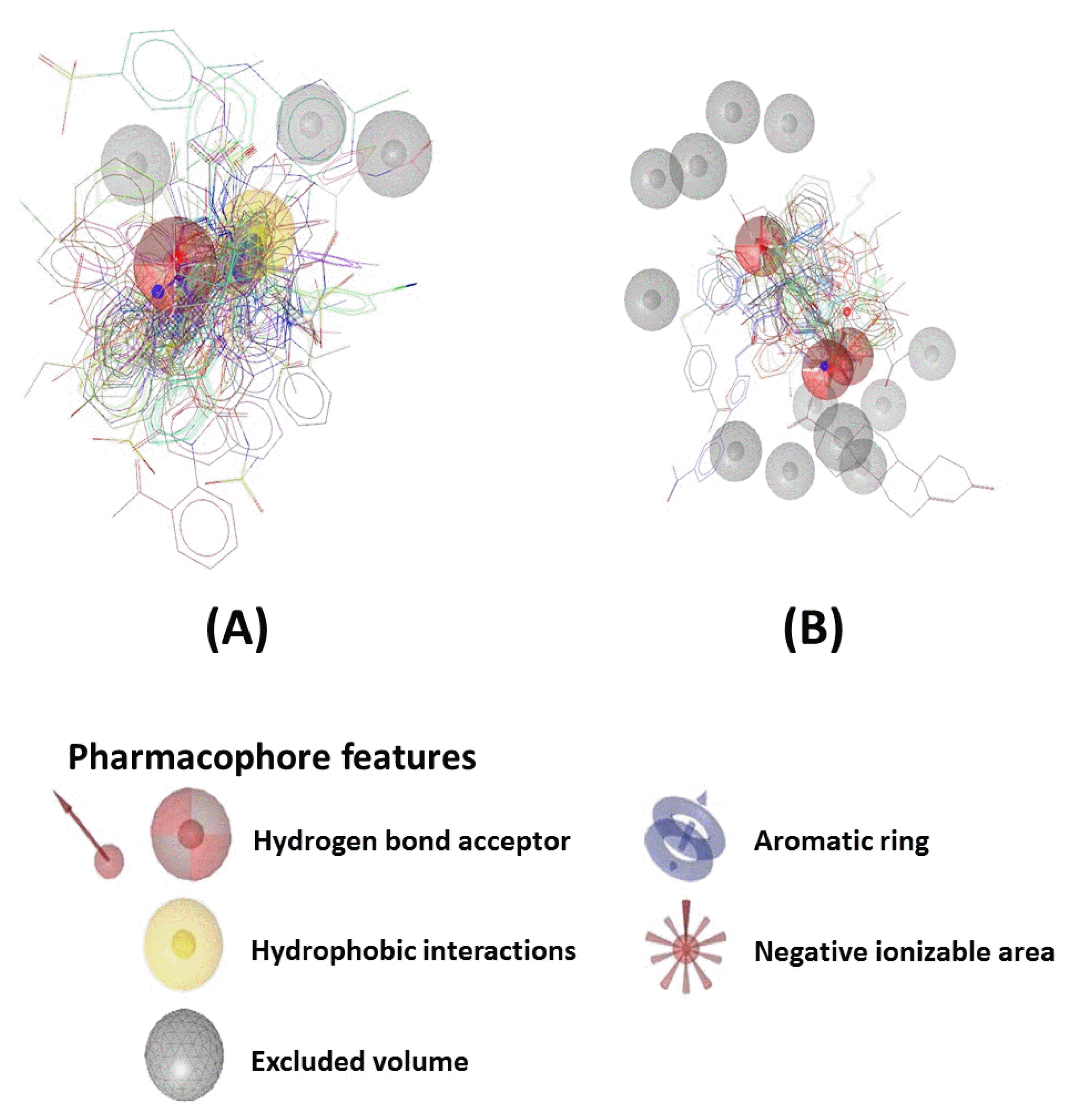
A total of 84 compounds from the ChEMBL database were first collected to generate a pharmacophore, but their structures and properties were too heterogeneous to get common features. Thus, we carried out clustering and categorized the compounds into two groups (Figs. 2,–4). For each group of compounds, we generated the corresponding pharmacophore model. Pharmacophore model 1 was generated by 49 compounds from group 1. The model was composed of four features (one hydrophobic centroid, one aromatic ring, two hydrogen acceptors) and three exclusion volume spaces (Fig. 5A). Model 2 was generated by 33 compounds from group 2. The model was composed of four features (one negative ionizable and three hydrogen bond acceptors) and 12 exclusion volume spaces (Fig. 5B).
For 3,563,829 lead-like compounds retrieved from the ZINC database, we performed a pharmacophore screen based on pharmacophore models 1 and 2 independently. Among multiple subsets provided by ZINC, we chose the lead-like subset, not the drug-like set, because we aimed to provide a list of potential hits that could be optimized further by other groups, as well as our group.
As a result, 400,153 and 290,742 hits fulfilled the features of models 1 and 2, respectively. The intersection of the two lists of hits, which fulfilled all features of both models, consisted of 38,087 compounds. To remove structurally similar compounds, we clustered the 38,087 hits by hierarchical clustering, based on the Tanimoto distance in PubChem Fingerprint. According to the result of the clustering, we ruled out redundant compounds that had similar compounds (Tanimoto coefficient >0.8). Thus, 1,338 hits eventually remained as potential inhibitors of APE1.
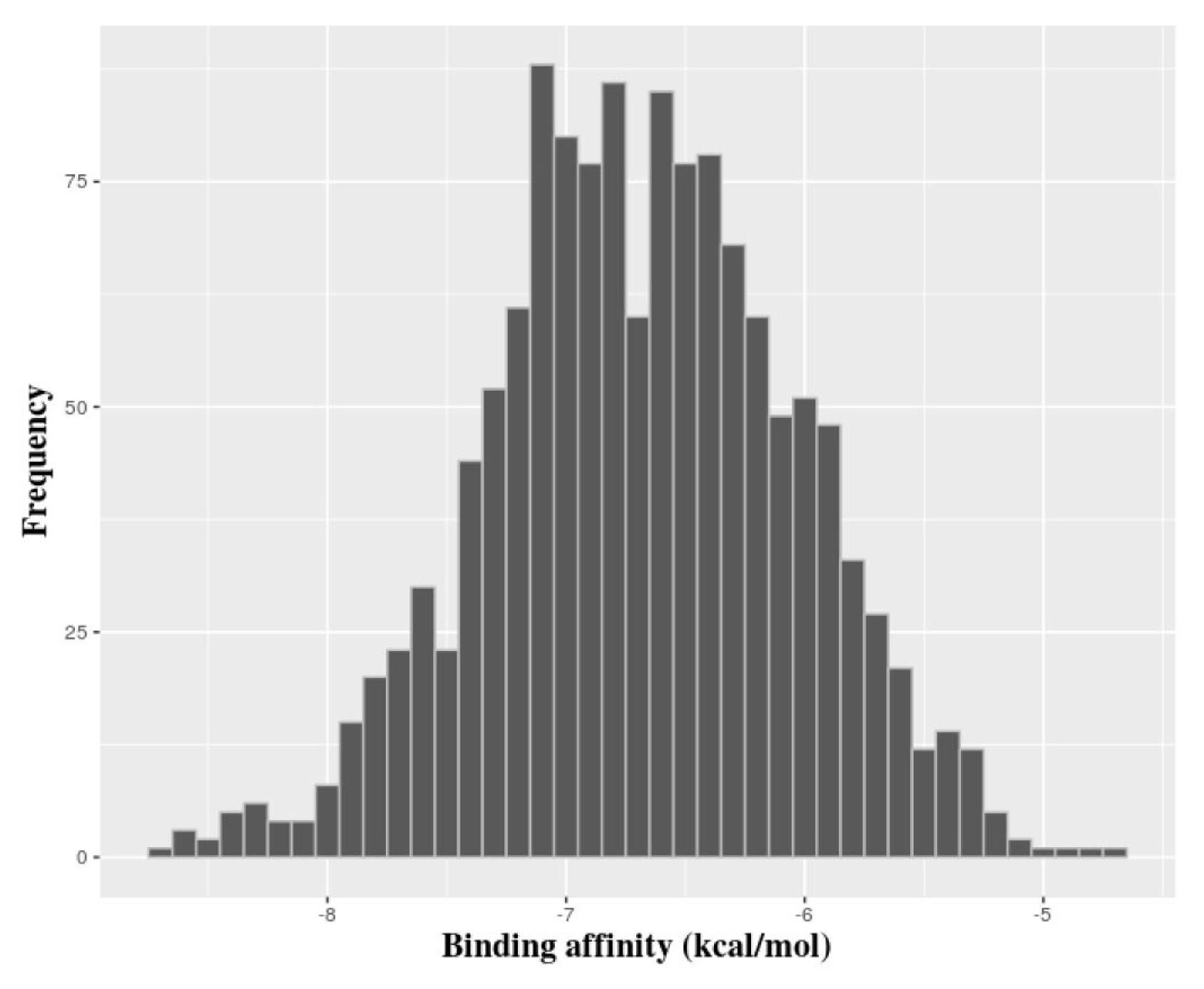
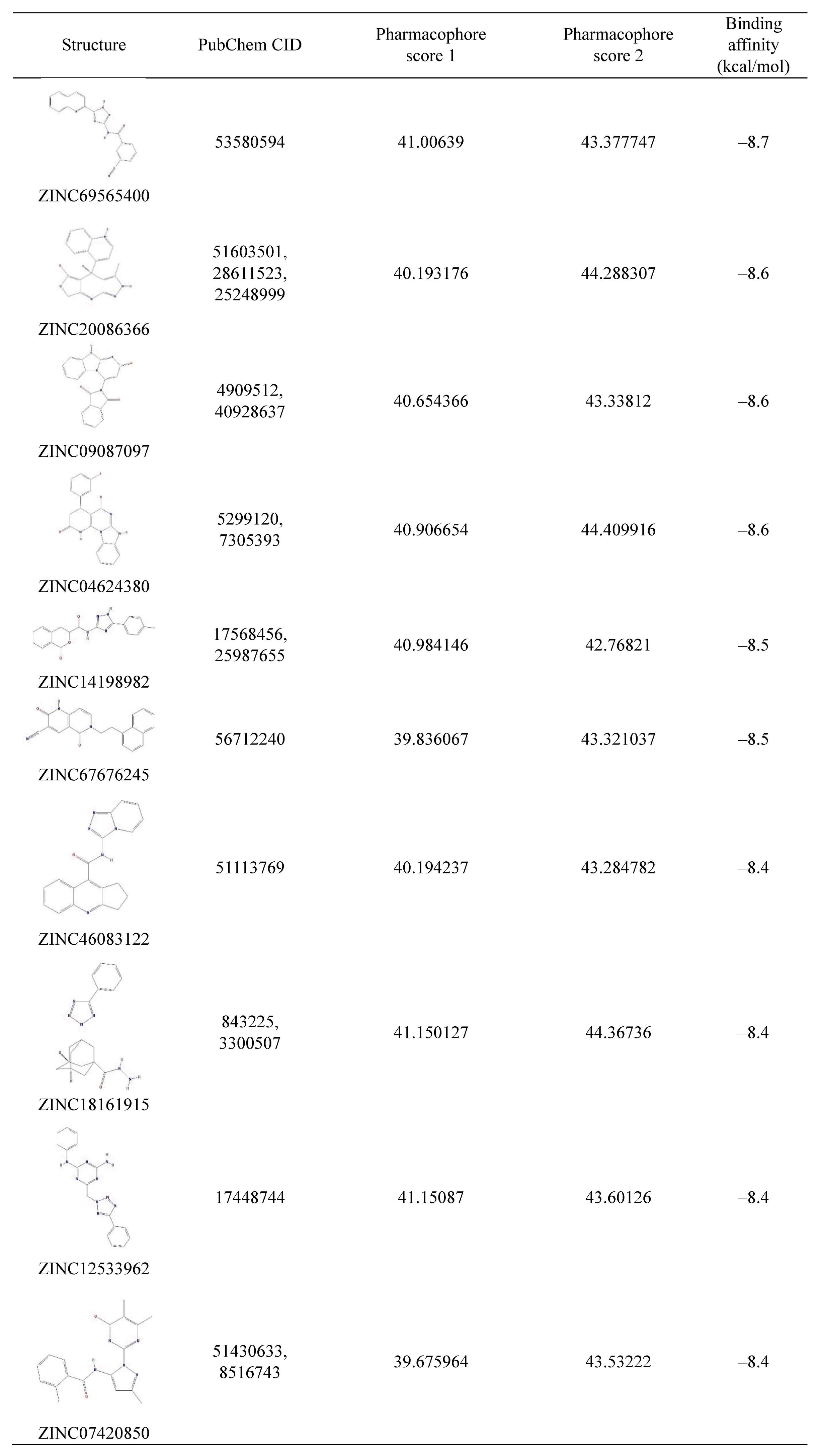
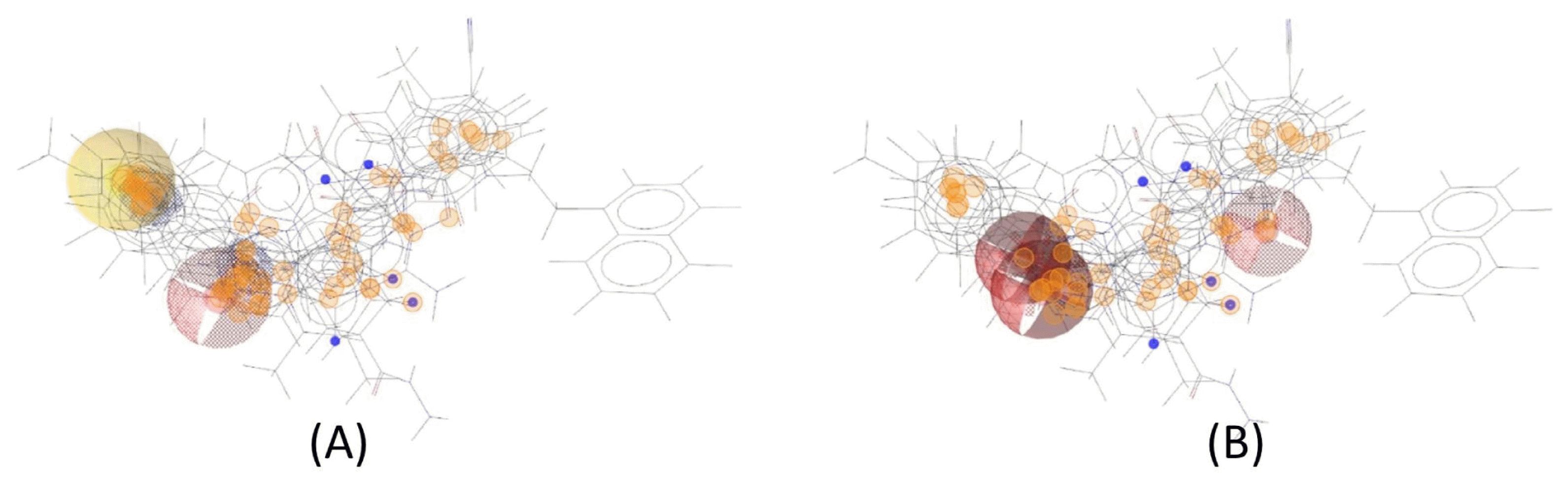
We carried out molecular docking of the hits against APE1 to prioritize the hits using AutoDock Vina. Fig. 6 depicts the distribution of the predicted binding energies of the hits of the pharmacophore screen by docking. After molecular docking, we did not filter out compounds based on a particular threshold of the predicted value of the binding affinity but instead provide the top 10 hits in Fig. 7, their predicted binding poses in Supplementary Fig. 1, and all of the hits in Supplementary Table 1. This is because although Shityakov and Förster [25] reported that a compound having a binding affinity predicted by AutoDock Vina of lower than −6 kcal/mol could be considered an active hit, the values are only predictive and rely on a somewhat empirical energy function. In other words, predicted binding affinities should be used restrictedly to help those who want to validate hits to determine the priority of subjects of an assay. Fig. 8 shows the alignments of the best hits into each pharmacophore model; all of the hits map well to the pharmacophore models. Of note, the rank of the docking results does not mean pharmacophore fitness, and all of the inhibitor compounds we found here can be mapped to the models well. The figure of pharmacophore alignment was made to provide an example showing that our hits can be mapped properly.
In summary, we screened more than 3 million lead-like compounds by pharmacophore modeling, and 1,338 hits were suggested to be potential inhibitors of APE1. However, this work has a limitation, due to the lack of experimental validation. Nevertheless, the list of hits in this work could reduce the time and cost of researchers who want to develop novel anticancer drugs inhibiting the activity of APE1, since we prioritized candidates of the experiments and since all of them have lead-like properties, which means that the hits are appropriate for further optimization and development into drugs.
Currently, there are several approaches that apply hits from a pharmacophore screen for further development in to a novel drug. Fei et al. [26] first developed a pharmacophore model of a drug target, like our method; then, 3D-quantitative structure-activity relationship (QSAR) modeling was used for validation and further virtual screening. Wieder et al. [27] proposed a novel approach combining pharmacophore modeling and molecular dynamics (MD) simulations, and they showed that their methods were likely to result in more robust hits. Like these approaches, the results from pharmacophore modeling could be adopted in other in silico methods, such as molecular docking, QSAR modeling, and MD simulation. It is worth combining these methods and our results to get more robust results. If further integrative approaches and in vitro or vivo assays of hits validate our results, our method could be applied to other drug targets, in addition to APE1.