Introduction
Human insulin, produced by beta-cells of the pancreatic islets, plays a critical role in the regulation of the metabolism of glucose [1]. Dysfunction of the synthesis or release of insulin may lead to diabetes mellitus [2]. Millions of people suffer from diabetes mellitus worldwide [3], and the most common medication to treat diabetes is insulin; so, a large number of studies on insulin have been done [4]. Insulin is first produced as an inactive protein, called preproinsulin. Preproinsulin, including a signal peptide, is a single, long protein. The chain evolves into proinsulin by cutting out the signal peptide. Then, proinsulin needs to be cleaved into insulin (an A chain and B chain) by removing the C-peptide linking the two chains [5].
Nowadays, recombinant DNA techniques enable us to produce insulin through biochemical processes using Escherichia coli [6]. However, the production of proinsulin by E. coli strains has to several drawbacks, such as low expression, difficulty in solubilizing the inclusion body, short half-life in the host cell, high proteolysis, and inefficient translation of the underlying coding sequences [7]. A new fusion protein system, which fuses an artificial leader peptide to the N-terminus of proinsulin, has been invented as a solution to these problems [8]. The specificity of cleavage and refolding rates are known to be dependent on the sequences of leader peptides, and different kinds of sequences have been proposed [6, 7, 9–11]. Thus, how the sequence of the artificial leader peptide affects the production of proinsulin and its modification into mature insulin needs to be investigated.
In this work, we used several kinds of computational approaches to find a better leader peptide. First, we predicted the three-dimensional structures of fused proinsulins while changing the leader peptides. Although the 3D structure of proinsulin was determined recently [12] and although that of active insulin was determined long ago [13], the structure of fused proinsulin should be analyzed by prediction. Afterwards, the stabilities of the predicted structures were calculated by molecular dynamics (MD) simulation. MD is a method that simulates the movements of atoms and calculates their potential energies. Finally, the interaction of proinsulins with protease was evaluated by protein-protein docking. By comparing these structural analyses and experiments, we demonstrate that these structural analyses may contribute to determining whether a leader peptide results in efficient production of insulin.
Methods
Selection of artificial leader peptides
In previous work (patent WO 2004/044206 A1), a formula for constructing a leader peptide was proposed as follows: Met-Thr-Met-Ile-Thr···Lys(Arg). We selected 13 models, as shown in Table 1. Out of these 13 models, 5 have been used to generate experimental results in a previous work [7], and 7 are being introduced in this work.
Protein structure prediction
The full sequence (leader peptide of the model plus the native sequence of proinsulin) of each fused protein was used as an input for the structure prediction. Among various methods of 3D protein structure prediction, we used I-TASSER (Iterative Threading ASSEmbly Refinement) [14], which ranked as first in performance in recent community-based competitions [15, 16].
MD simulation
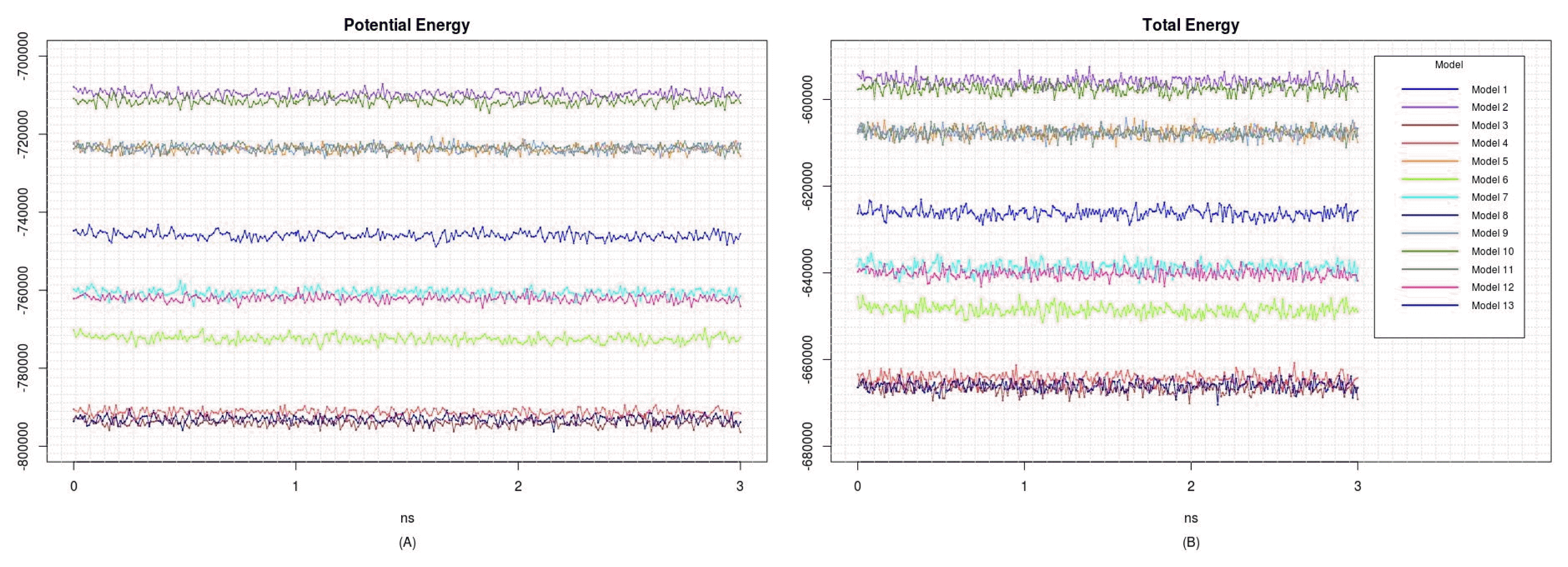
The predicted structures were located in a cubic box of water; then, 3-ns MD simulations were carried out using GROMACS, ver. 5.1.2 [17]. Amber99sb was chosen as the force field [18]. Energies of the models were minimized using the steepest descent algorithm for 3 ns. The step size was 0.002 ps. The properties and stabilities of the models were evaluated based on the potential energy, total energy, and root-mean-square deviation of the atomic positions.
Protein-protein docking
Protein-protein docking between trypsin (PDB ID: 2PTN) and recombinant proinsulins was performed using the InterEvDock server [19]. The InterEvDock server provides three kinds of scores: InterEvDock, FRODOCK [20], and SOAP_PP [21]. We chose the SOAP_PP score to evaluate the binding affinities, and docked poses were used to check whether two proteins were bound at the right position.
Results and Discussion
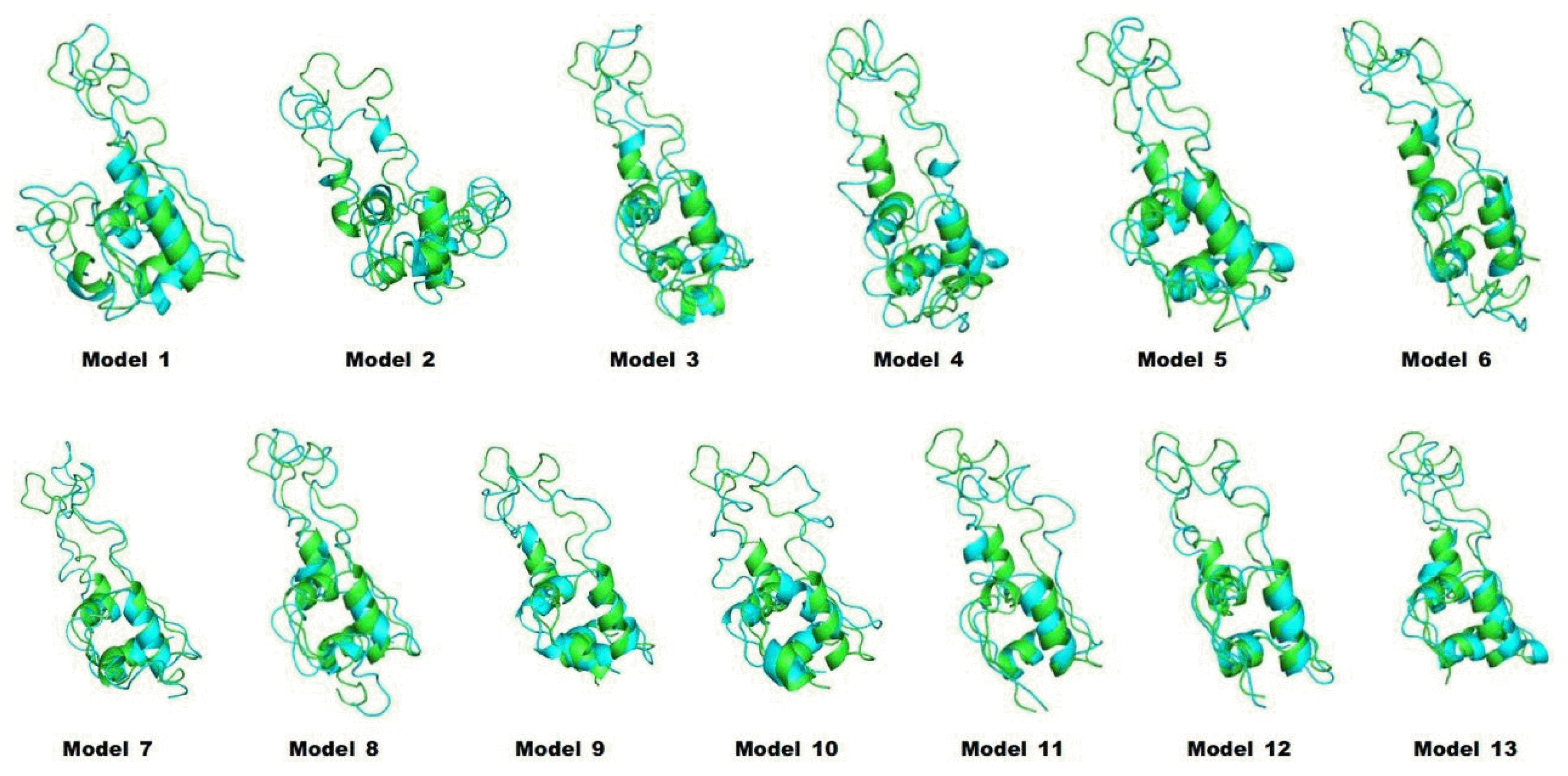
We predicted the 3D structures of 13 artificial models of proinsulin using I-TASSER (Fig. 1). As shown in Fig. 1, different leader peptides affected the overall structure of proinsulin. Especially, although the sequences of models 1 and 2 had only a 1-residue difference, the predicted positions of the leader peptides were quite different.
To refine the predicted structures, we observed the changes in structures by MD simulation at 3 ns to minimize energy. The structures after the MD simulation were superimposed onto the original structures by I-TASSER (Fig. 2). All potential energies of models were kept relatively stable (Fig. 3). The predicted structures were little refined; thus, they were naturally favorable. The total energy and potential energy of models 3 and 8 were the lowest. Among the models tested before (Table 1), no correlation between refolding yields and energies was observed.
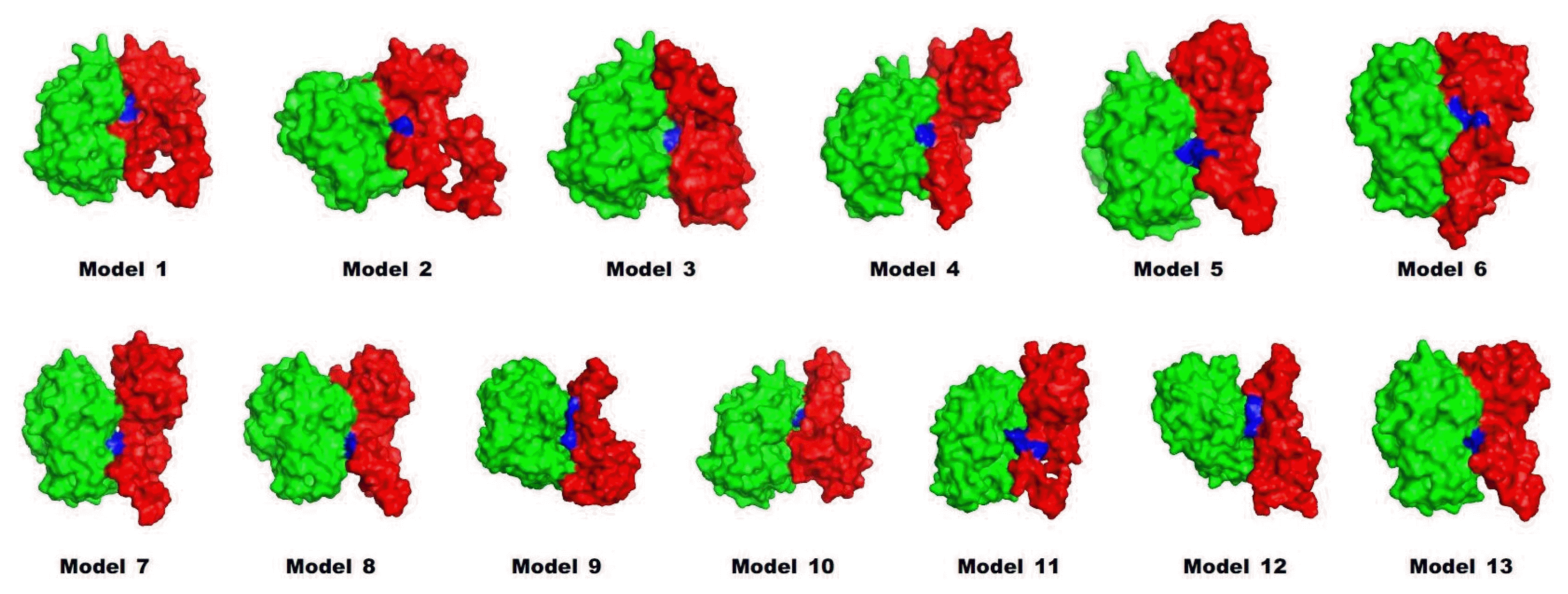
Preproinsulin should be refolded and cleaved to become mature insulin. A previous work showed that trypsin could be used as an efficient protease in the maturation of insulin [7]. To investigate how the leader peptide affects the binding mode between proinsulin and trypsin, in silico docking was carried out by InterEVDock [19].
Interestingly, in the docked structure (Fig. 4), the active sites (histidine 57, aspartate 102, and serine 195) of trypsin were located near lysine 64 and arginine 65 of proinsulin, which is the exact position where the first cleavage of proinsulin occurs to release chain A. Binding affinities were also predicted by InterEVDock (Table 2). Among all docked structures, models 1 and 2 were predicted to have the strongest binding affinities.
Because multiple steps of cleavage are needed for maturation, we performed additional protein docking of trypsin to model structures whose C-terminal chain after residue 64 (A chain) was cleaved out. As a result of this step, the preferable position for the active sites of trypsin were located near arginine 31 and arginine 32, which are known to form a cleavage site between the B and C chains. The binding affinities of the models were predicted to be strong (in descending order): model 2, 3, and 1. The two docking steps for artificial proinsulin models into trypsin revealed leader peptides that did not affect the order of cleavage sites, and models 1 and 2 are likely to be best accessed by trypsin in producing mature insulin. This coincides with previously tested refolding yields [7].
In summary, using 13 artificial models of leader peptides of proinsulin, we predicted the structures of fused proinsulins, the structures were refined by MD simulation, and protein-protein docking revealed binding modes between the artificial models and trypsin. The energies of the predicted structures of the models were not related to refolding yields, but the docking energies between the protease and the models showed some relation. We expect these analyses to provide basic information in a structural context for more effective production of insulin.