Perspectives of Integrative Cancer Genomics in Next Generation Sequencing Era
Article information
Abstract
The explosive development of genomics technologies including microarrays and next generation sequencing (NGS) has provided comprehensive maps of cancer genomes, including the expression of mRNAs and microRNAs, DNA copy numbers, sequence variations, and epigenetic changes. These genome-wide profiles of the genetic aberrations could reveal the candidates for diagnostic and/or prognostic biomarkers as well as mechanistic insights into tumor development and progression. Recent efforts to establish the huge cancer genome compendium and integrative omics analyses, so-called "integromics", have extended our understanding on the cancer genome, showing its daunting complexity and heterogeneity. However, the challenges of the structured integration, sharing, and interpretation of the big omics data still remain to be resolved. Here, we review several issues raised in cancer omics data analysis, including NGS, focusing particularly on the study design and analysis strategies. This might be helpful to understand the current trends and strategies of the rapidly evolving cancer genomics research.
Introduction
In the last decade, numerous genomic studies have addressed the enormous complexity of the cancer genome. Genomic profiling using microarray technology could stratify the tumors into homogeneous subgroups, providing novel clinical insights for the development of diagnostics and therapeutics as well as systematic views on the underlying mechanisms of tumor progression. In addition to the microarray technologies, explosive advances on sequencing technologies have been made recently, which is called "next generation sequencing (NGS)." Compared to the previous DNA sequencing of the Sanger method using dideoxynucleotide termination reaction termed as "first-generation" sequencing, NGS uses massively parallel sequencing method generating hundreds of millions of short (~200 bp) DNA reads, which can sequence a human genome rapidly with extremely lower cost. The earlier NGS method with the single-end read sequencing inevitably produces the short-read problems, limiting the accuracy of genome alignment. This could be improved by applying a paired-end sequencing method, allowing substantial advances in identifying not only point mutations but also genomic rearrangements, such as deletions, amplifications, inversions, translocations, and gene-fusions [1, 2].
The NGS technology is now divided into "second generation sequencing" and "third generation sequencing." The second generation sequencing refers to the strategies of short-read alignment, while the rapidly being developed technology of the third generation sequencing refers to the single DNA molecule based sequencing. The third generation method has advantage of less amount of DNA input that allows the emerging field of single cell sequencing [3]. Moreover, there is no step for PCR amplification, therefore, the nucleotide incorporation errors can be handled.
However, all the platforms of NGS technologies still have limitations in accurate base calling and alignment. The errors are likely to be platform-dependent, which increases the complexity of the data analysis. Therefore, the cost for bioinformatic analysis, rather than the sequencing itself continues to grow, which is referred to as "the $1,000 genomes, the $100,000 analysis" problem [4].
Cancer Genome Analysis with NGS Technologies
The application of Exome-Seq and Whole Genome-Seq profiled a mutational spectrum in various cancers [5-8]. The RNA-Seq could profile not only the gene expressions but also new parameters such as allelic expression, alternative splicing [9, 10], RNA-editing [11], and alternative polyadenylation of 3'-untranaslated region (3'-UTR) [12, 13]. Structural variations such as gene-fusions, e.g., TMPRSS2-ERG in prostate cancer [14] and KIF5B-RET in lung cancer [15] have been identified from the analysis of NGS data. Moreover, the patterns of genome rearrangements can be analyzed systematically. For example, a novel pattern of genomic rearrangement such as fold-back inversion could be found by simply examining the short read alignment of NGS data [16]. A novel mechanism of cancer genome rearrangement i.e., chromothripsis has been proposed, which represent a catastrophic event of fragmentation and reassembly of a single chromosome [17]. In addition, high-resolution epigenomic profiles of cancer genome could be obtained by applying NGS to chromatin immunoprecipitation-sequencing (ChIP-Seq) and methylated DNA immunoprecipitation-sequencing (MeDIP-Seq). For example, a recent study of the genome-wide DNA methylation profile showed novel patterns in which the majority of DNA methylation changes occurs at CpG island shores neighboring the regions up to 2 Kb from CpG islands, and revealed the patterns of cancer-specific and tissue-specific DNA methylation [18, 19]. Aberrant histone modification was also found in cancers showing an association with the patient's prognosis [20]. These studies support the pivotal role of epigenetic regulation in cancer development and progression. Moving forward, new applications such as genome-wide translocation sequencing (HTGTS) [21] and translocation capture sequencing (TC-Seq) [22] have recently been proposed, which could profile the genome-wide translocation hotspots.
Notably, the advantage of NGS is not restricted to these applications. It serves a platform for the identification of novel RNAs or DNAs. For example, the long noncoding RNAs (lncRNAs) that are transcribed from intergenic and intronic regions have been identified in prostate cancers i.e., prostate cancer-associated ncRNA transcripts (PCATs) [23]. A novel class of DNA i.e., extrachromosomal microDNA, has been recently found by NGS technology, which is derived from unique non-repetitive sequence enriched in 5'-UTR, exons, and CpG islands [24]. The role of microDNA in cancer will be the next question.
Challenges in cancer genomics
The primary study goals of cancer genomics are not simple, which include the studies to get either clinical or mechanistic insights from the cancer genome. To clarify the complicated study designs and strategies of cancer genomics, we have categorized the gene signatures obtained from cancer genome data into four classes prediction, phenotype, function, and molecular targets based on the study goals [25]. The majority of the previous studies have suggested the translational or clinical utility of the genomics data by addressing the candidate biomarkers or the prediction signatures for predicting patients clinical outcomes, such as recurrence, survival, metastasis, or response to therapies. Notwithstanding the overwhelming identification of candidate biomarkers from the cancer genome, only a handful of candidate biomarkers that have been discovered from genomic analyses can succeed in the validation of the clinical utility [18]. There are several challenges in cancer genomics that preclude clinical utility. One of them would be data reproducibility. They might be due in part to the experimental biases as well as sample cohort issues. The use of different platforms measuring gene expressions and different data processing methods could produce biased observation in each study. Increasing sample size will be one of the solutions to find proper biomarkers, overcoming the reproducibility problem. Undoubtedly, large-scale sample collection provides increased statistical power. However, previous studies, even with large sample sizes, have often failed to reproduce their findings in independent studies [26]. This might be due mostly to the use of different protocols and analysis methods. Moreover, biased sample collection may also affect the performance of prognostic biomarkers, leading to subsequent failure to validate the biomarker in another patient population [27]. For example, diagnostic biomarkers must be discovered in early-stage tumors; however, the sample collection of early tumors with enough of a sample size might be difficult in the clinical setting [18, 28]. In addition, the cost-effectiveness of the sample size enrolled in a study should be considered. Simply increasing the sample size might not be the best solution.
The sample sources and qualities are also important factors to be considered in the study design. For example, circulating DNAs or microRNAs in the plasma or urine can be used to develop "noninvasive" biomarkers in cancer patients, which might bring the technology much closer to the clinic [29, 30]. Attempts to use formalin-fixed paraffin-embedded (FFPE) tissues might also be more applicable to the clinic [31], although the quality and the quantity of the DNAs or RNA extracts from FFPE or plasma are still problematic for genomics studies requesting further elaboration.
Dissecting the tumor heterogeneity
The data complexity comes not only from the heterogeneous or biased sample composition, but also from the innate complexity of tumor biology. Previously, mounting evidence has shown the enormous heterogeneity of tumors at the molecular level. The tumor heterogeneity can be explained by two hypothetical models. One is the clonal segregation model with a multi-step process, and the other is the cancer stem cell theory. The cancer stem cell model describes the heterogeneous cellular origin of cancers from primitive progenitor cells to mature differentiated cells, which may contribute to tumor heterogeneity. In this context, genomic profiling studies could define the cancer subpopulation harboring stem-like traits in various cancer types, supporting the cancer stem cell theory [32, 33]. Similarly, we also defined the bilineal trait in a subpopulation of hepatocellular carcinoma (HCC) by comparing the gene expression profiles of HCC and cholangiocarcinoma (CC) [34]. This result showed the continuous liver cancer spectrum between HCC and CC, suggesting that stem-like or de-differentiation traits may give rise to the heterogeneous progression of HCC. We also suggested that the dysfunction of p53 machinery is associated with the acquisition of the stemness trait in HCC [35]. Recently, this association could be validated by showing p53 knockout mouse model can give rise to bilineal liver cancers [36].
In addition, various host factors may contribute to tumor heterogeneity. Interactions of the tumor cells with host eco-system, such as innate immune systems, or the reactions of surrounding microenvironment against the tumor may affect the tumor behaviors [37]. Thus, the proper detection of biomarkers might be difficult without considering the effect of host factors. Furthermore, the intratumoral heterogeneity of cancers have been notified in detail by genome-wide sequencing of multi-loci from the same tumor [38]. The comparison of primary and metastatic tumors by single cell sequencing also revealed the sequential mutation process during cancer progression [39]. Similarly, the comparison of mutations from multiple HCC tumors in the same patient could define the evolutionary lineage among tumors cells [40]. Strikingly, the development of single-cell sequencing technology could provide a more detailed and systematic view on intra-tumoral heterogeneity [41, 42]. Of interest, such attempts enabled the construction of phylogenic trees from mutational heterogeneity, which revealed evolutionary tumor growth opening a new field of "cancer evolution".
Returning to biology for clinical utility
As discussed above, there are many factors contributing to tumor heterogeneity, which may impede the discovery of new biomarkers. Thus, we are now urgently in need of developing new strategies for biomarker discovery from cancer genome data. Considering the huge complexity of the cancer genome and the limitation of current technologies, it would be a reliable strategy to evaluate the functional relevance of the candidate biomarkers rather than simply showing the statistical significance of the association by enrolling larger samples or applying more stringent statistics. Although the conventional strategies for biomarker discovery do not require the functional significance of the candidates [43], current hurdles in cancer genomics request a functional validation step in the pipeline of the biomarker discovery. Our limited understanding of the complexity of cancer biology is a significant challenge for translational interpretation of the cancer genome.
Challenges of big data issues and integromics
More recently, systematic structuring and integration of multiple and multi-layered omics data resources, i.e. integromics, are thought of a state-of-the-art strategy. The recent establishment of The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) could accelerate and facilitate the integration and sharing of cancer genome data [44]. Multi-layered integromics could define tumor heterogeneity, revealing pivotal aberrations of genetic events or signaling pathways [45]. In parallel, genomic repositories for drug activities have been established [46-48]. Linking the profiles of drug sensitivity to the cancer genome could provide a powerful platform to guide rational and personalized cancer therapeutics.
Now, as genomic data are increasing and accumulating enormously, new study designs and analysis strategies for integromics might be required, particularly with the context of tumor heterogeneity and the discovery of the functional biomarkers. As shown in Fig. 1, the first step in cancer integromics is the dissection of tumor heterogeneity. Then, the next step will be a recapitulation of the relations between clinical/biological phenotypes and molecular genotypes in cancer subpopulations, which can address novel functionalities in particular subpopulations of cancers. We suggest that the discovery of reliable candidate functional biomarkers as well as functional genetic alterations, so-called "driver events", can be achieved through performing this step-by-step evaluation of both clinical and functional utilities. This hybrid study design would open an exciting era for developing new "functional biomarkers" and preventive/therapeutic strategies with the consideration of biological backgrounds of tumor heterogeneity.
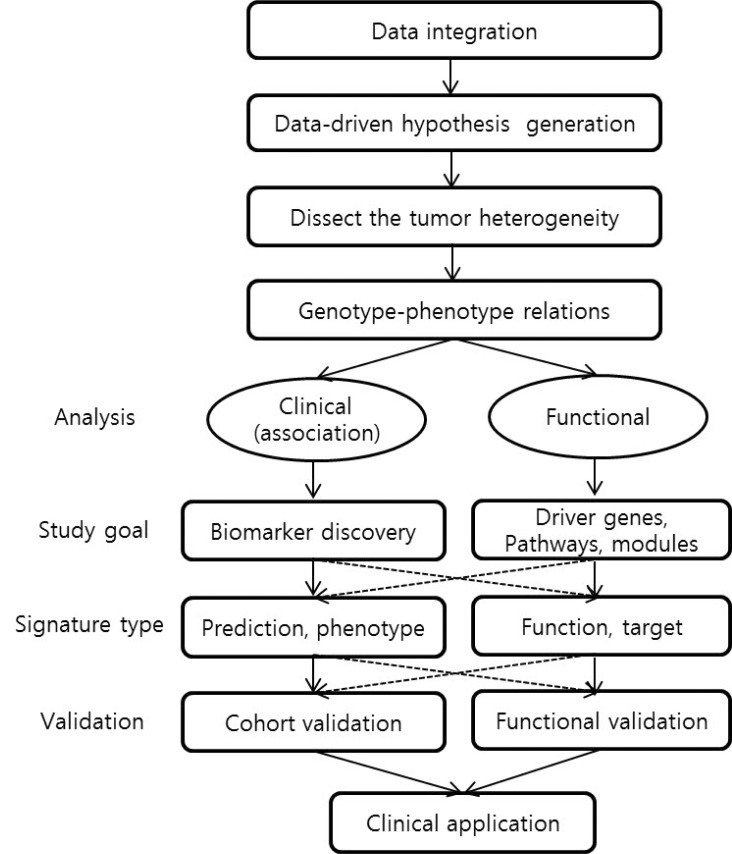
Hybrid study design for integromics approach.
Conclusion
We briefly reviewed the current challenges and perspectives of integrative cancer genomics, focusing particularly on the complexity of omics data and cancer biology. Integromic approaches with functional evaluation should be considered even for clinical as well as mechanistic applications of cancer genome data. Necessarily, the challenges of big data (particularly the NGS platforms) and integromics should be considered in the study design. A deep understanding of both cancer biology and omics data characteristics is necessarily required for successful cancer genome analysis. Moving forward, it is clear that progress will come through large-scale, wide-scope, and multi-disciplinary collaborations and sharing systems, which will accelerate the realization of translational and personalized medicine in the near future.
Acknowledgments
This work was supported by grants of the Korea Healthcare Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111574).