A Fosmid Cloning Strategy for Detecting the Widest Possible Spectrum of Microbes from the International Space Station Drinking Water System
Article information
Abstract
In this study, fosmid cloning strategies were used to assess the microbial populations in water from the International Space Station (ISS) drinking water system (henceforth referred to as Prebiocide and Tank A water samples). The goals of this study were: to compare the sensitivity of the fosmid cloning strategy with that of traditional culture-based and 16S rRNA-based approaches and to detect the widest possible spectrum of microbial populations during the water purification process. Initially, microbes could not be cultivated, and conventional PCR failed to amplify 16S rDNA fragments from these low biomass samples. Therefore, randomly primed rolling-circle amplification was used to amplify any DNA that might be present in the samples, followed by size selection by using pulsed-field gel electrophoresis. The amplified high-molecular-weight DNA from both samples was cloned into fosmid vectors. Several hundred clones were randomly selected for sequencing, followed by Blastn/Blastx searches. Sequences encoding specific genes from Burkholderia, a species abundant in the soil and groundwater, were found in both samples. Bradyrhizobium and Mesorhizobium, which belong to rhizobia, a large community of nitrogen fixers often found in association with plant roots, were present in the Prebiocide samples. Ralstonia, which is prevalent in soils with a high heavy metal content, was detected in the Tank A samples. The detection of many unidentified sequences suggests the presence of potentially novel microbial fingerprints. The bacterial diversity detected in this pilot study using a fosmid vector approach was higher than that detected by conventional 16S rRNA gene sequencing.
Introduction
Rationing and water recycling are an essential part of life at International Space Stations (ISSs) [1]. Water purification machines on the ISS partly mimic these processes, but they do not rely on microbes or any other living organisms. These machines cleanse wastewater in a 3-step process. The first step involves a filter that removes particles and debris. The water then passes through multifiltration beds that contain substances that remove organic and inorganic impurities. Finally, the catalytic oxidation reactor removes volatile organic compounds and kills bacteria and viruses. Once the water is purified, astronauts try to ensure that the water is used with maximum possible efficiency. Even with intense conservation and recycling efforts, the space station will gradually lose water because of inefficiencies in the life support system.
Water that passes through animals' bodies is refreshed by natural processes [2]. Microbes in the soil break down urea and convert it to a form that plants can absorb and use to build new plant tissue. Water excreted by animals also evaporates into the atmosphere and rains back down to the earth as freshwater-a natural form of distillation. ISSs always need to be resupplied with water, because no water reprocessing technology is currently available for use in space. National Aeronautics and Space Administration (NASA) scientists continue to look for ways to improve the life-support systems of space stations, which can thereby reduce water losses and aid in finding ways to reuse other waste products. Bacteria and viruses are extraordinarily abundant in water, soil, and sediment microbial ecosystems [3]. In drinking water, soil, and sediment, viral abundance exceeds that of co-existing host populations by 10- to 100-fold [4]. Microscopy and molecular genetic tools have been critical in demonstrating that bacteria and viruses are dynamic components of microbial ecosystems capable of significantly influencing the productivity and population biology of their host communities. Moreover, these approaches have begun to describe the immense genetic diversity of viral communities. A critical first step in the application of many cultivation-independent approaches to virus ecology is obtaining a viral concentrate from an environmental sample. Culture-dependent methods also rely on viruses being present at sufficiently high levels to be detected [3]. In this study, the methodology for the isolation and concentration of viruses from water, soil, and aquatic sediment samples has been discussed in detail.
Microorganisms constitute two-thirds of the earth's biological diversity [5, 6]. As many as 99% of the microbes present in certain environments can not be detected by regular techniques [7]. Protocols have been developed to assess unexplored microbial diversity in order to overcome the existing barriers that prevent the estimation of biodiversity [8]. New screening methods have been designed to construct specific fosmid libraries that can help distinguish novel microbial populations in drinking water samples [9]. To screen the complete populations, cloning vectors, like fosmids and bacterial artificial chromosomes (BACs), are being developed [10-12]. This article describes the various methodologies and tools that have been developed to understand and identify the widest spectrum of microbial communities, including bacteria, archaea, and viruses, through fosmid cloning analysis.
Bacteriophage phi29 (Φ29) DNA polymerase, the 66-kD product of viral gene 2, has been shown to be capable of efficient DNA amplification both in vitro and in vivo [13-15]. This enzyme is very reliable with respect to the amplification of genomic DNA for constructing libraries, especially when the starting genomic material is limited. This enzyme requires 2 proteins for its activity: viral DNA polymerase P2 and Φ29 terminal protein p3 [16]. The enzyme has high processivity (>70 kb) and a remarkable strand-displacement ability with single-stranded DNA and double-stranded DNA. Based on this enzymatic characteristic, the replication of both DNA strands can be processed from each terminal-priming event without the synthesis of RNA-primed Okazaki fragments. The enzyme also has a 3'-5' proofreading exonuclease function [17].
Bacteriophage ϕDNA polymerase amplifies circular DNA in a rolling-circle amplification (RCA) mechanism. This characteristic has been used to amplify and clone the complete circular DNA genome of bacteria and viruses. The amplified DNA could be used for direct sequencing and was cloned after digestion with a single cutting restriction endonuclease. The use of this enzyme simplified the cloning steps and increased the cloning efficiency of the complete genome of circular DNA viruses [18]. The Φ29 DNA polymerase was ideal for our project because of the limited amount of starting material available. In addition, the microbes in our sample could not be cultivated using traditional approaches. Using exonuclease-resistant random primers, Dean and colleagues have achieved up to a 10,000-fold amplification from 1 ng of M13 template and have also improved the average product length to greater than 40 kb [19]. The method is based on multiple displacement amplification technology and provides high yields of approximately 40 µg of DNA from only a few genome copies.
In a previous study on subsurface flow-constructed wetlands, PCR-based methods were developed and used for the detection of pathogenic microbial populations, such as Campylobacter spp. and Yersinia enterocolitica, in treated wastewater [20, 21]. In this study, we constructed fosmid libraries to assess the microbial diversity in drinking groundwater samples from Kennedy Space Center's ground service equipment tank and the space shuttle water tank (iodine-treated water) and examined the samples for the presence of many different bacterial populations during the water purification processes. The goals of this study were: 1) to compare the sensitivity of the fosmid cloning strategy with that of traditional culture-based and 16S rRNA-based approaches and 2) to detect the widest possible spectrum of microbial populations during the water purification process.
Methods
Collection of water samples
Two drinking water samples from the Kennedy Space Center, Orlando, Florida, USA-namely, water samples from the ground service equipment tank (Prebiocide) and the space shuttle water tank (iodine-treated water; Tank A)-were collected for microbial diversity assessment.
Isolation of genomic DNA
Approximately 500 mL each of the Prebiocide and Tank A water samples was concentrated using several Amicon Ultra-15 filters (50K NMWL; Millipore, Billerica, MA, USA). The filtered concentrates were combined together and further concentrated to 100 µL by using a vacuum evaporator (HT-4 II Evaporation system; Genevac Inc., Valley Cottage, NY, USA) at 28℃ for 2 h. DNA isolation was conducted using Trizol reagent, following the instructions provided by the manufacturer (Invitrogen, Carlsbad, CA, USA). During the last step, the DNA pellets were dissolved in 10 µL of 8 mM NaOH and then incubated at 60℃ for 10 min.
Amplification of genomic DNA
The purified DNA was subjected to randomly primed RCA to amplify any DNA present in the water samples, according to the procedure detailed in the GenomiPhi DNA Amplification Kit manual (Amersham Biosciences, Piscataway, NJ, USA). This kit consists of 3 components: the sample buffer; the reaction buffer; and an enzyme mix containing random primers, dNTPs, and Φ29 DNA polymerase [22]. One microliter of the concentrated water sample from either the Prebiocide or Tank A was denatured with 9 µL of sample buffer at 95℃ for 5 min and cooled rapidly on ice. The amplification reaction was performed by adding 9 µL of reaction buffer and 1 µL of enzyme mix to the denatured sample. The reaction mixtures were incubated at 30℃ for 16 h. After amplification, the tubes were incubated at 65℃ for 10 min to inactivate the Φ29 DNA polymerase enzyme. The amplified genomic DNA was precipitated by ethanol and then dissolved in 10 mM Tris-HCl (pH 7.5) and 1 mM ethylenediaminetetraacetic acid. Most of the random primers and dNTPs were removed after precipitation with ethanol.
Construction of the fosmid library
The amplified high-molecular-weight DNA from the Prebiocide or Tank A water samples was cloned with fosmid vectors. In order to select DNA fragments longer than 20 kb, the amplified DNA was visualized by pulsed-field gel electrophoresis (PFGE) in 1% low-melting-point agarose at 4.5 V/cm in 1 × TBE buffer for 13 h. DNA fragments larger than 20 kb were cut out, extracted from the gel, and re-run for 5 h under the same conditions. These resolved DNA fragments were used to construct a fosmid library by following the procedure detailed by the manufacturer. Using the CopyControl Fosmid Library Construction Kit (Epicentre, Madison, WI, USA), the sheared DNA was end-repaired to generate blunt 5'-phosphorylated ends and then ligated directly into the cloning-ready CopyControl pCC1FOS vector (Fig. 1) at a 10:1 vector:insert ratio. The ligated DNA was then packaged using ultrahigh-efficiency MaxPlax Lambda Packaging Extract (Epicentre) and was plated on phage T1-resistant Escherichia coli cells to produce the library. Packaging a single 10-µL ligation reaction generated more than 100,000 fosmid clones from the DNA isolated from the Prebiocide and Tank A water samples. DNA from several randomly chosen clones was purified using the Direct Lysis Fosmid96 Kit (Epicentre) for high-throughput fosmid sequencing.

CopyControl pCC1FOS Fosmid vector map. The pCC1FOS cloning vector was linearized at its Eco72 I (blunt) site, dephosphorylated, and used for cloning blunt-end genomic DNA. CMR, chloramphenicol resistance.
Restriction enzyme analysis
One microliter of the RCA product from the Prebiocide or the Tank A samples was incubated with 10 U of HindIII at 37℃ overnight. Restriction digestion products were separated by 1% agarose gel electrophoresis and visualized by ethidium bromide staining. In addition, among the 9,740 clones obtained from the fosmid library constructs, subsets of randomly selected clones from the Prebiocide or Tank A samples were digested with NotI restriction enzyme for measuring insert DNA sizes.
DNA sequencing
Each colony from the different libraries was picked and then inoculated in Luria Broth media containing chloramphenicol and glycerol (10%, final concentration) in a 384-well plate. The cells were cultured at 37℃ for 1 day and stored at -80℃. DNA was isolated using the Qiagen Miniprep Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Initially, 100 clones from each library were selected for the sequencing reaction (Macrogen, Seoul, Korea). BLAST sequence homology searches were conducted against the GenBank database after subtracting the vector sequence. Subsequently, 1,260 colonies from the Prebiocide library and 2,600 colonies from the Tank A library were sequenced. The major sequences encoding specific genes were identified from both Prebiocide and Tank A samples on the basis of protein sequences (with more than 75 amino acid residues) from GenBank.
PCR detection of different microbial groups in drinking water samples
DNA was extracted from the Prebiocide and Tank A water samples by using the Qiagen Miniprep Kit. Several microbial groups were analyzed by PCR amplification of gene fragments of several phylogenetic groups representing different microbial communities [23]. These groups included general bacteria, Bacillus species, gram-positive bacteria (high G + C content), Streptomyces species, eukaryotes (fungi, protists, and green algae), and general fungi. The PCR primer pairs used in the analysis and the groups they represent are listed in Table 1.

Primer pairs used for PCR amplification to detect the microbial diversity in the Prebiocide and Tank A drinking water samples
Results
Genomic DNA amplification using the Phi29 DNA polymerase
The DNA plug did not show any detectable amount of DNA when analyzed using PFGE. The GenomiPhi DNA Amplification Kit (Amersham Biosciences) is claimed to have very high sensitivity for DNA detection; the smallest trace of DNA should be amplified if present. In the protocol provided with the amplification kit, it is recommended that the amplification reaction be started with 10 ng of DNA. Although the results initially showed that the concentrated samples did not contain any detectable amount of DNA, the amplification procedure was continued with these samples. The control (DNase free water) and the DNA isolated from the Prebiocide and Tank A samples showed a similarly positive pattern after DNA amplification, which seemed to be nonspecific due to the contamination of the Φ29 DNA polymerase enzyme with exogenous DNA (Fig. 2A). Similarly, the positive controls, such as lambda DNA, showed significant amplification of up to 20-30 µg in a 20-µL reaction. The distribution of the amplified DNA ranged from 20 to 40 kb. Repeated trials of less than 3 h did not result in any amplification; however, a DNA product was noted after 6 h of incubation. The size of the amplified DNA (between 20 kb and 40 kb) was determined by PFGE.
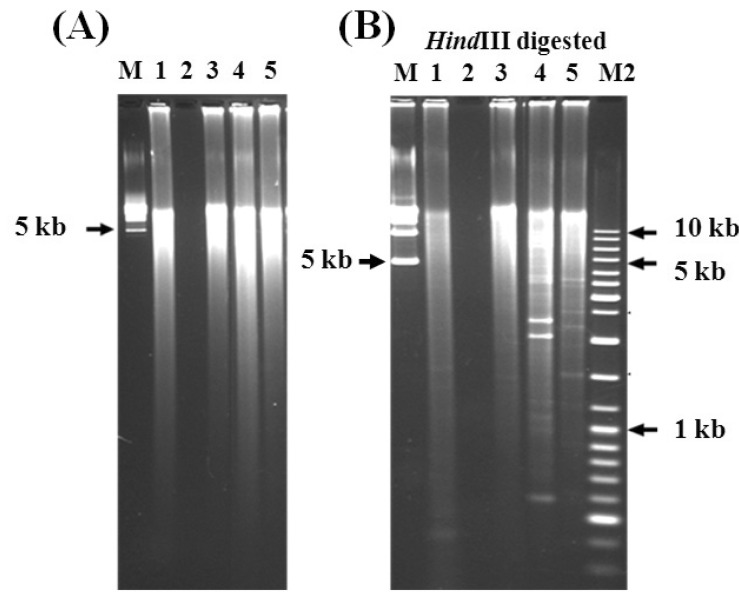
DNA gel pictures. (A) Genomic DNA amplification by rolling-circle amplification for Prebiocide and Tank A drinking water samples. Lane M, 5 kb DNA ladder; lane 1, Prebiocide DNA product; lane 2, negative control (no Φ29 DNA polymerase); lane 3, Tank A DNA product; lane 4, positive control (lambda DNA 10 ng); lane 5, control (DNase-free water). Amplification conditions: 6 h, 30℃. The amplified DNA was analyzed by 1% agarose gel electrophoresis in 0.5× TBE buffer at 150 V. (B) Restriction digestion of the amplified genomic DNAs by using HindIII enzyme. Lanes are same as in (A). Lane M2, 1 kb DNA ladder. The digested samples were analyzed by performing routine 0.8% agarose gel electrophoresis in 1× TBE buffer at 150 V.
The GenomiPhi DNA Amplification Kit manual explains that nonspecific amplification usually occurs, even in the absence of template DNA, because of the presence of random primers and the high amplification performance capability of the enzyme. In order to confirm whether the amplified DNA had been specifically amplified from the genomic DNA template, 1 µg of each of the amplified DNA samples was digested with HindIII (Fig. 2B). Several fragmented DNA bands with expected sizes were observed in the digestion reaction of the amplified DNA from the lambda (positive control), Prebiocide, Tank A, and control samples, showing that the amplification was specific. Smeared bands of indigestible DNA were also obtained because of partial and/or nonspecific amplification of the genomic DNA.
Construction of fosmid vector libraries
Before constructing the fosmid vector libraries, several domain or species-specific PCR reactions were performed. Both the Prebiocide and Tank A samples and the control (DNase-free water) showed amplification with different primer sets (Fig. 3). Primer pairs used for species-specific PCR amplification to detect the microbial diversity are listed in Table 1 [24-26]. After constructing the fosmid vector libraries, 4,000 clones from the Prebiocide sample, 2,290 clones from the Tank A sample, and 3,450 clones from the control were picked. As expected, NotI digestion of the fosmid DNA released and produced inserts that were more than 40 kb in size (Fig. 4). Subsets digested with the NotI enzyme were sequenced. Only DNA sequences that exhibited more than 80% similarity with an existing GenBank sequence were included in the analysis.
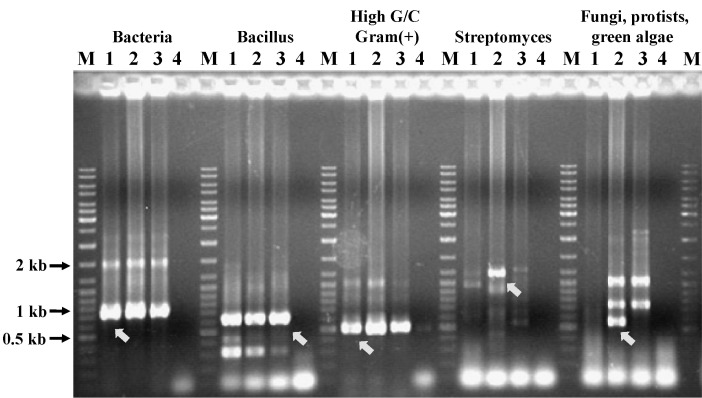
Genomic DNA amplification and detection of different microbial groups by PCR in drinking water samples. Lane M, DNA ladder; lane 1, amplified Prebiocide DNA product; lane 2, amplified Tank A DNA product; lane 3, amplified control DNA product; lane 4, no DNA input for PCR. The amplified DNA product was analyzed by 1% agarose gel electrophoresis in 0.5× TBE buffer at 150 V. The arrowhead shows the amplification of genomic DNA and the presence of corresponding microbial DNA.
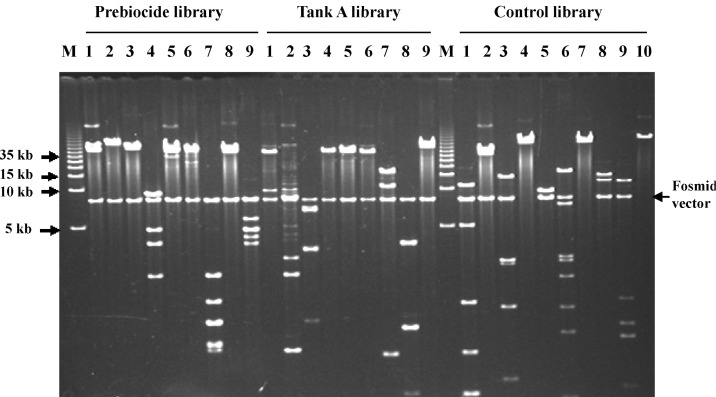
NotI restriction enzyme digestion. A combination of NotI restriction enzyme digestion of fosmid DNA isolated from randomly chosen clones from the Prebiocide, Tank A, and control (DNase free water) libraries and ethidium bromide-stained agarose gel electrophoresis verifies the presence of a >40-kb insert. Lane M, DNA ladder; lanes 1-10 show NotI-digested fosmid DNA selected from the Prebiocide, Tank A, and control libraries. Pulsed-field gel electrophoresis conditions: 4.5 V/cm, 1-7 s, 13 h, 1× TBE buffer at 14℃.
DNA sequence analysis
BLAST sequence homology searches were also performed against the GenBank database after subtracting the vector sequence. Subsequently, 1,260 single colonies from the Prebiocide library and 2,600 single colonies from the Tank A library were picked. The major sequences encoding specific genes were then identified from the Prebiocide (26 clones), Tank A (27 clones), and control samples (21 clones) on the basis of the GenBank protein sequences. Supplementary Table 1 shows representative BLAST search results from GenBank for the Prebiocide, Tank A, and control sample clones.
Identification of microbial populations and diversity
The microbial populations, identified on the basis of sequences for both the Prebiocide and Tank A drinking water samples, are listed in Table 2 and Supplementary Table 1. Bradyrhizobium japonicum, Mesorhizobium loti, and Bradyrhizobium species were present in the Prebiocide library. Bradyrhizobium and Mesorhizobium belong to rhizobia, a large community of nitrogen fixers often found in association with plant roots [27]. Ralstonia metallidurans and R. eutropha were detected in Tank A. Strains of R. metallidurans (renamed as Cupriavidus metallidurans strain CH34) are prevalent in soils with high heavy metal content [28]. Sequences encoding specific genes from Burkholderia fungorum, a species abundant in soil and groundwater, were found in both the Prebiocide and Tank A samples [23, 29]. Protein sequences homologous to those of several genera of opportunistic pathogens were retrieved by RCA. The genomic DNA amplification by PCR detected different microbial groups in drinking water samples (Table 1, Fig. 3).

List of microbial populations identified on the basis of sequences in both the Prebiocide and Tank A drinking water samples
Discussion
In the current study, a fosmid cloning strategy was used to assess the microbial populations in the ISS drinking water system-namely, samples from the Prebiocide and Tank A. Initially, no microbes could be cultivated, and conventional PCR reactions failed to amplify 16S rDNA fragments from low biomass samples. Therefore, randomly primed RCA was used to amplify any DNA present in the samples, followed by size selection using PFGE. Genomic DNA amplification was performed using the DNA isolated from concentrated DNA samples obtained from Prebiocide and Tank A drinking water samples in the ISS.
We used the GenomiPhi DNA Amplification Kit from Amersham; the DNA amplification experiments using a Φ29 DNA polymerase proved to be ideal for the scarce amount of genomic starting material available. However, nonspecific amplification can occur either in the presence or in the absence of the DNA template in the reaction. For the lambda DNA sample, the presence of amplified DNA can be confirmed if the Φ29 DNA polymerase is specific to the template DNA by digestion with restriction enzymes. Lambda DNA (48.5 kb) has many HindIII restriction sites, producing a unique distribution of sizes. However, even in the case of lambda DNA, we confirmed that some amount of indigestible amplified DNA still exists, which appears to be due to nonspecific amplification and/or partial amplification (Fig. 2B). According to the Amersham kit user manual, the enzyme synthesizes single-stranded DNA or double-stranded DNA. An alternative explanation is that there were many single-stranded DNA fragments that could not be cut by restriction enzymes. To test this theory, the amplified DNA was denatured at 95℃ for 3 min and reannealed at room temperature for 30 min to decrease the amount of single-stranded DNA. However, restriction digestion still resulted in a large amount of indigestible DNA (data not shown).
Using the concentrated water samples also proved that such samples had sufficient DNA, which could be used to amplify genomic DNA by using commercially available kits, even when only a few microbes were present. Nevertheless, amplification of genomic DNA may require some additional DNA preparation steps, because boiling cells for a short time may not effectively release the genomic DNA for use as template. We decreased the incidence of nonspecific amplification by reducing the incubation time to less than 12 h when it was possible to use the amplified DNA as a template.
The amplified high-molecular-weight DNA obtained from the Prebiocide, Tank A, and control samples was cloned into fosmid vectors. Several hundred clones were randomly selected for sequencing, which was followed by Blastn/Blastx searches (Table 2, Supplementary Table 1). Sequences encoding specific genes from Burkholderia, a species abundant in soil and groundwater, were obtained for both the Prebiocide and Tank A samples [23, 29]. Bradyrhizobium and Mesorhizobium, belonging to rhizobia, a large community of nitrogen fixers often found in association with plant roots, were present in the Prebiocide library [27]. Ralstonia, which is prevalent in soils with a high heavy metal content, was detected in the Tank A library [28]. Giardia lamblia is a common cause of diarrhea in humans, and the presence of homologous protein sequences with the protein encoded by G. lamblia in Tank A samples warrants further study [30].
The current NASA technologies used for treating drinking water in the ISS are effective in removing viable cultivable microbes. Conventional PCR failed to detect any amplifiable rRNA genes, which suggests that the purification of this drinking water was satisfactory. However, the amplification of naked DNA from these samples enabled elucidation of the microbial diversity in the samples. When drinking water was not treated with a biocide, protein sequences from diverse genera could be retrieved. The detection of many unidentified sequences (data not shown) also suggests the presence of potentially novel microbial fingerprints. The bacterial diversity observed in this pilot study with the fosmid approach was greater than that observed with conventional 16S rRNA gene sequencing.
Acknowledgments
This work was funded by an Advanced Environmental and Monitoring and Control Program of NASA grant, under contract with the California Institute of Technology. This work was also partly supported by the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (2012016803) and the Priority Research Centers Program (NRF 2012-0006687). We thank Darrell Jan for his constant encouragement and Randy Sumner for collecting the water samples.
References
Supplementary materials
Supplementary data including one table can be found with this article online at http://genominfo.org/src/sm/gni-10-249-s001.pdf