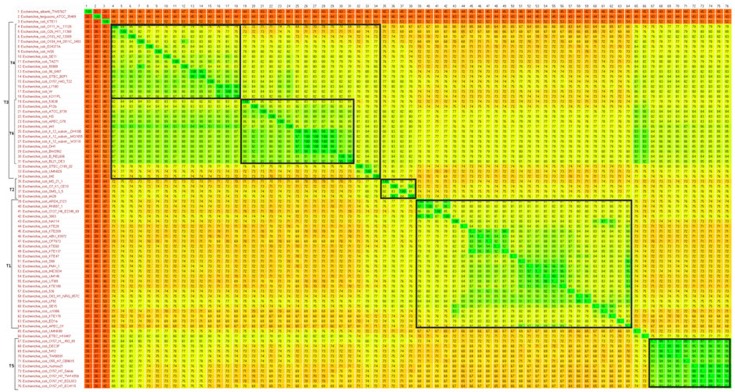
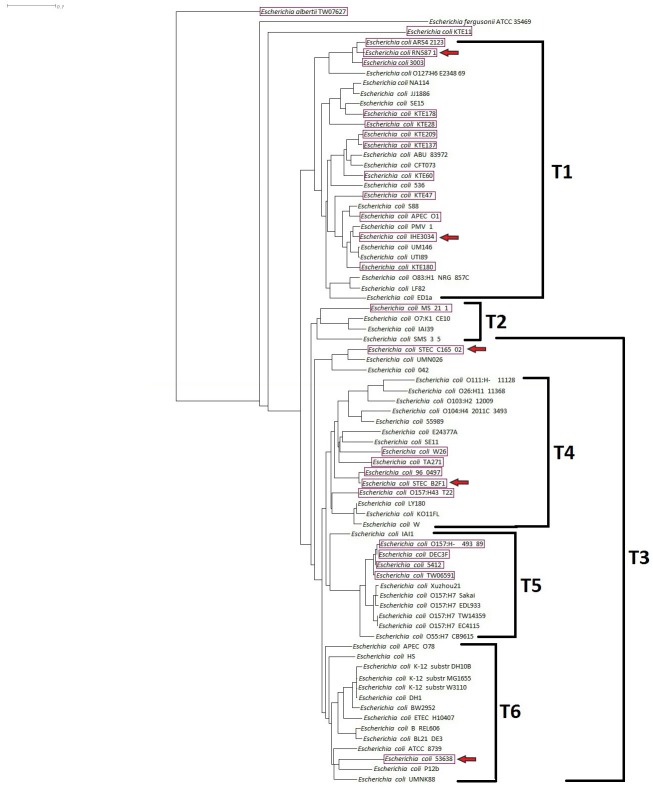
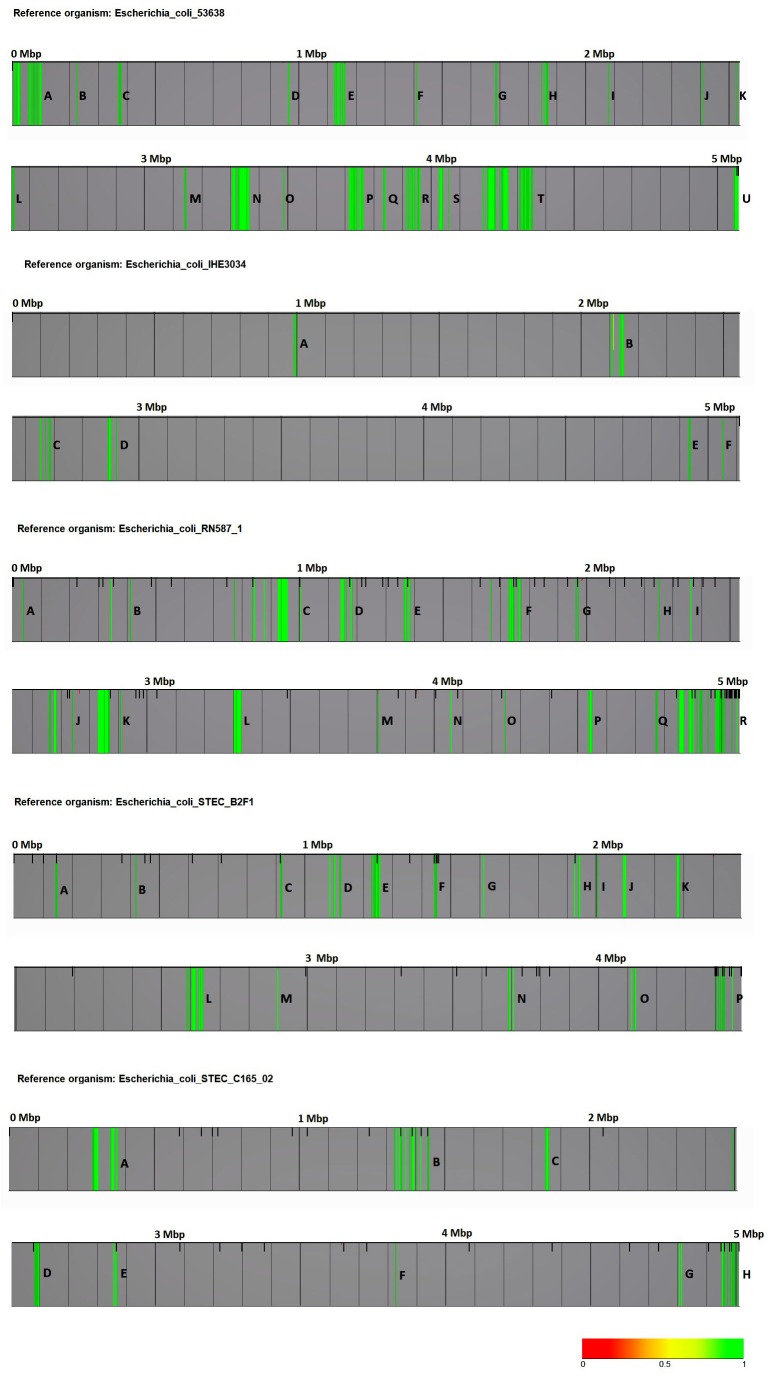
|
Cytolethal distending toxin |
Cytolethal distending toxin A |
Cytolethal distending toxin, subunit C |
Cytolethal distending toxin A/C family protein |
Cytolethal distending toxin C |
Cytolethal distending toxin A/C family protein |
|
Cytolethal distending toxin B |
Cytolethal distending toxin, subunit B |
Cytolethal distending toxin A/C family protein |
|
Cytolethal distending toxin subunit C |
Cytolethal distending toxin, subunit A |
|
Holin |
Phage holin, lambda family |
Holin, lambda family |
Holing |
-a
|
Phage holin, lambda family |
|
Nuclease |
Exodeoxyribonuclease 8 |
Exonuclease family protein |
Exonuclease family protein |
Endonuclease/Exonucl ease/phosphatase family protein |
Restriction endonuclease family protein |
|
Hypothetical protein ECRN5871_4153, [HNH endonuclease family protein] |
Type I site-specific deoxyribonuclease, HsdR family |
Type I site-specific deoxyribonuclease, HsdR family protein |
|
Hypothetical protein ECSTECC16502_028 0, [HNH endonuclease] |
|
Endonuclease/Exonucl ease/phosphatase family protein |
|
Phage integrase |
Phage integrase |
Integrase/recombinase, phage integrase family |
Integrase |
Phage integrase family protein |
Integrase |
|
Prophage integrase |
Site-specific recombinase, phage integrase family |
Phage integrase family protein |
Prophage lambda integrase |
Prophage CP4-57 integrase |
|
Integrase for prophage CP-933T |
Prophage lambda integrase |
|
Integrase domain protein |
|
Putative membrane protein |
Putative membrane protein |
Hypothetical protein ECOK1_2122, [membrane protein] |
Outer membrane autotransporter barrel domain protein |
Putative membrane protein |
Putative membrane protein |
|
Hypothetical protein Ec53638_1156, [membrane protein] |
Hypothetical protein ECOK1_2557, |
Hypothetical protein ECSTECB2F1_3192, [membrane protein] |
|
OmpA-like transmembrane domain protein |
|
Outer membrane porin protein LC |
|
Outer membrane protein lom |
|
Regulatory proteins |
Phage regulatory protein Cro |
Putative transcriptional regulator DicA157 |
Regulatory protein CII |
Transcriptional regulator, AraC family |
4-Hydroxyphenylaceta te catabolism regulatory protein HpaA |
|
Transcriptional regulator, AlpA family |
Putative regulatory protein Cox |
Prophage CP4-57 regulatory protein family protein |
Prophage CP4-57 regulatory protein family protein |
|
Putative phage regulatory protein, Rha family |
Transcriptional regulator, LacI family |
|
Restriction-modification system |
Putative type I restriction-modification system, S subunit |
-a
|
Type II restriction enzyme EcoRII |
Type I restriction modification DNA specificity domain protein |
Type I restriction-modification system specificity determinant |
|
Type I restriction-modification system specificity subunit |
Modification methylase EcoRII |
Type III restriction enzyme, res subunit |
|
Type I restriction-modification enzyme, R subunit |
Type I restriction enzyme specificity protein |
|
Type I restriction-modification system, M subunit |
Type I restriction-modification system, M subunit |
|
Tail fiber assembly family, baseplate assembly proteins, Tail fiber protein and Tail tape measure protein |
Tail fiber assembly protein |
Tail fiber protein |
Caudovirales tail fiber |
Tail fiber assembly |
Caudovirales tail fiber assembly family protein |
|
Phage P2 baseplate assembly protein gpV |
Phage tail tape measure protein |
Assembly family protein |
Hypothetical protein ECSTECB2F1_0901, [tail fiber assembly protein, caudovirales tail fiber assembly protein] |
Tail fiber |
|
Hypothetical protein ECRN5871_3504,[tail fiber assembly protein] |
Tail fiber domain protein |
|
Putative tail fiber protein |
Baseplate assembly protein V, W |
Caudovirales tail fiber assembly family protein |
Phage tail fiber repeat family protein |
|
Tail fiber |
Long tail fiber protein p37 domain protein |
Prophage tail fiber family protein |
|
Phage tail tape measure protein family |
Tail fiber domain protein |
Phage tail fiber repeat family protein |
|
Phage tail tape measure protein, TP901 family, core region |
Phage tail tape measure protein, lambda family |
|
Terminase |
Phage terminase large subunit |
-a
|
Phage small terminase subunit |
Phage terminase large subunit family protein |
Terminase small subunit |
|
Terminase |
Terminase, ATPase subunit |
Terminase B protein domain protein |
|
Terminase, endonuclease subunit |
Terminase B protein |
|
Terminase large subunit |
|
Terminase small subunit |
|
Transferase |
Pyruvyl transferase |
-a
|
Hypothetical protein |
Putative teichuronic acid biosynthesis glycosyltransferase tuaG |
Acetyltransferase family protein |
|
Glycosyl transferase domain protein, group 2 family |
ECRN5871_3051, [nucleotidyl transferase, PF08843 family] |
Glucose-1-phosphate thymidylyltransferase |
Hypothetical protein ECSTECC16502_1 295, [acetyltransferase] |
|
Glycosyltransferase, sugar-binding region containing |
D12 class N6 adeninespecific DNA methyltransferase family protein |
RTX toxin acyltransferase family protein |
|
DXD motif |
Hypothetical protein ECRN5871_0025, [N-acetyltransferase CN5] |
Acetyl-CoA acetyltransferase |