Mutational Analysis of Extranodal NK/T-Cell Lymphoma Using Targeted Sequencing with a Comprehensive Cancer Panel
Article information
Abstract
Extranodal natural killer (NK)/T-cell lymphoma, nasal type (NKTCL), is a malignant disorder of cytotoxic lymphocytes of NK or T cells. It is an aggressive neoplasm with a very poor prognosis. Although extranodal NKTCL reportedly has a strong association with Epstein-Barr virus, the molecular pathogenesis of NKTCL has been unexplored. The recent technological advancements in next-generation sequencing (NGS) have made DNA sequencing cost- and time-effective, with more reliable results. Using the Ion Proton Comprehensive Cancer Panel, we sequenced 409 cancer-related genes to identify somatic mutations in five NKTCL tissue samples. The sequencing analysis detected 25 mutations in 21 genes. Among them, KMT2D, a histone modification-related gene, was the most frequently mutated gene (four of the five cases). This result was consistent with recent NGS studies that have suggested KMT2D as a novel driver gene in NKTCL. Mutations were also found in ARID1A, a chromatin remodeling gene, and TP53, which also recurred in recent NGS studies. We also found mutations in 18 novel candidate genes, with molecular functions that were potentially implicated in cancer development. We suggest that these genes may result in multiple oncogenic events and may be used as potential bio-markers of NKTCL in the future.
Introduction
Extranodal natural killer (NK)/T-cell lymphoma, nasal type (NKTCL) is a malignant neoplasm characterized by a cytotoxic phenotype and is associated with Epstein-Barr virus (EBV) infection. It is rare in Western countries but is the most common subtype of mature T and NK cell neoplasms in Asia [1]. Although it has been suggested that EBV could play a pathogenic role in the development of NKTCL, its etiology and molecular pathogenesis have remained unexplained.
The clinical course of NKTCL is usually highly aggressive, but its survival rate has improved with the recent progress in intensive therapies, including combined chemoradiotherapy and autologous bone marrow transplantation [23]. However, the prognosis of NKTCL is variable, and the recurrence rate is still high. Thus, there is a great need to define a molecular pathway associated with the pathogenesis, prognosis, or drug resistance of NKTCL.
Next-generation sequencing (NGS) technology has made genetic and molecular studies effective, with more reliable results achieved [4]. High-throughput sequencing technology can conduct millions of sequencing reactions in parallel. Unlike in Sanger sequencing, NGS enables the detection of novel genetic alterations, since the sequence of every base in the region of interest is detected, and a large number of loci of interest (multiple genes) can be analyzed in a single panel. It is currently used in a wide variety of research fields. For example, it has been used to identify mutations or to profile gene expressions in cancer research and has been utilized in population genomic studies, including the 1000 Genomes Project.
Recent advancements in NGS technology have revolutionized clinical research. Moreover, the emergence of bench-top sequencers, such as the Ion Proton platform, has allowed genomic and molecular studies to be done easily and effectively by individual researchers and institutes. Nowadays, many molecular studies on the cancer genome are being performed using NGS. The detection of mutations or gene expression in mixed samples requires high sequencing depth, which is not achieved by whole-genome "shotgun" approaches. Targeted sequencing is able to yield a desirable sequencing depth for this purpose. Thus, it is more efficient to identify mutations correlating with tumor phenotype and in developing possible targeted drug therapies. Yet, molecular studies using NGS in NKTCL have languished compared with B-cell lymphoma cases [56789]. Therefore, this study aims to identify mutations possibly responsible for NKTCL using targeted sequencing with a comprehensive panel of 409 cancer-related genes (Supplementary Fig. 1) and to suggest candidate genes that can be used as potential bio-markers of NKTCL in the future.
Methods
Clinical information
Samples from tumors diagnosed between 2011 and 2014 were collected from Dankook University Hospital (Cheonan) in South Korea. Five formalin-fixed and paraffin-embedded (FFPE) tumor samples from NKTCL were analyzed. NKTCL was diagnosed according to the 2008 World Health Organization classification [10]. In all cases, immunohistochemical studies for CD3, CD56, and granzyme B, as well as EBV-encoded small RNA in situ hybridization, were carried out with the FFPE tissues. All immunohistochemical results were positive for the markers, but one case was negative for EBV (Supplementary Fig. 2). Patient ages ranged from 46 to 80 years. One case was from the submandibular region. The remainder was from the nasal cavity. Supplementary Table 1 summarizes the information of the enrolled subjects.
DNA preparation
FFPE tissue samples were deparaffinized in xylene, and 3-to 5-mm thick sections were extracted from the samples. Using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), we isolated genomic DNA from the samples following the manufacturer's instructions. The quality control results of the DNA preparations are illustrated in Supplementary Table 2 and Supplementary Fig. 3.
Library preparation and Ion Proton sequencing
Targeted gene sequencing was performed as previously described [11]. Ten nanograms of the DNA preparations was used as a template for multiplex polymerase chain reaction (PCR) of a 409-gene panel covering coding regions (Ion AmpliSeq Comprehensive Cancer Panel; Life Technologies, Grand Island, NY, USA).
Fragment libraries were constructed by DNA fragmentation, barcode and adaptor ligation, and library amplification using the Ion DNA Barcoding kit (Life Technologies) according to the manufacturer's instructions. The size distribution of the DNA fragments was analyzed using a bioanalyzer and the High Sensitivity kit (Agilent, Santa Clara, CA, USA). Using the Ion Xpress Template kit (Life Technologies), we performed template preparation, emulsion PCR, and Ion Sphere Particle (ISP) enrichment according to the manufacturer's instructions. The ISPs were loaded onto a P1 chip and sequenced using an Ion P1 sequencing kit (Life Technologies).
Variant calling and annotation
Ion Proton platform-specific pipeline software (Proton Suite v4.4; Life Technologies) was used to separate the barcoded reads, generate sequence alignments with the hg19 human genome reference, perform target-region coverage analysis, and filter poor signal reads. Initial variant calls were generated using Proton Suite with a plug-in program (variant caller v4.4). Variant calls were further analyzed using internally developed software that allows variant filtering and annotation using refGene in University of California Santa Cruz (UCSC), 1000 Genomes, COSMIC v.67, dbSNP build 138, and ExAC. To minimize false positives, variants were filtered with a normal population variant database, The Korean Personal Genome Project (http://opengenome.net/) [12].
Candidate variant detection (variant filtering)
To filter out false-positives, which were hardly regarded as true somatic mutations or disease driver mutations, we used several additional filtering steps that are generally accepted and generated the final variant calls, as shown in Fig. 1. They included (1) transition-type point mutation; (2) indel >2 bp; (3) normal variants of Inhouse, 1000 Genomes Project, and ExAC_est_Asia (Broad Institute); (4) homozygous-type mutation; (5) conservation <0.3; (6) regions except for missense, splice site, stop gained, and frameshift; (7) total depth <100; and (8) altered allele frequency >0.3 or <0.05 [13].
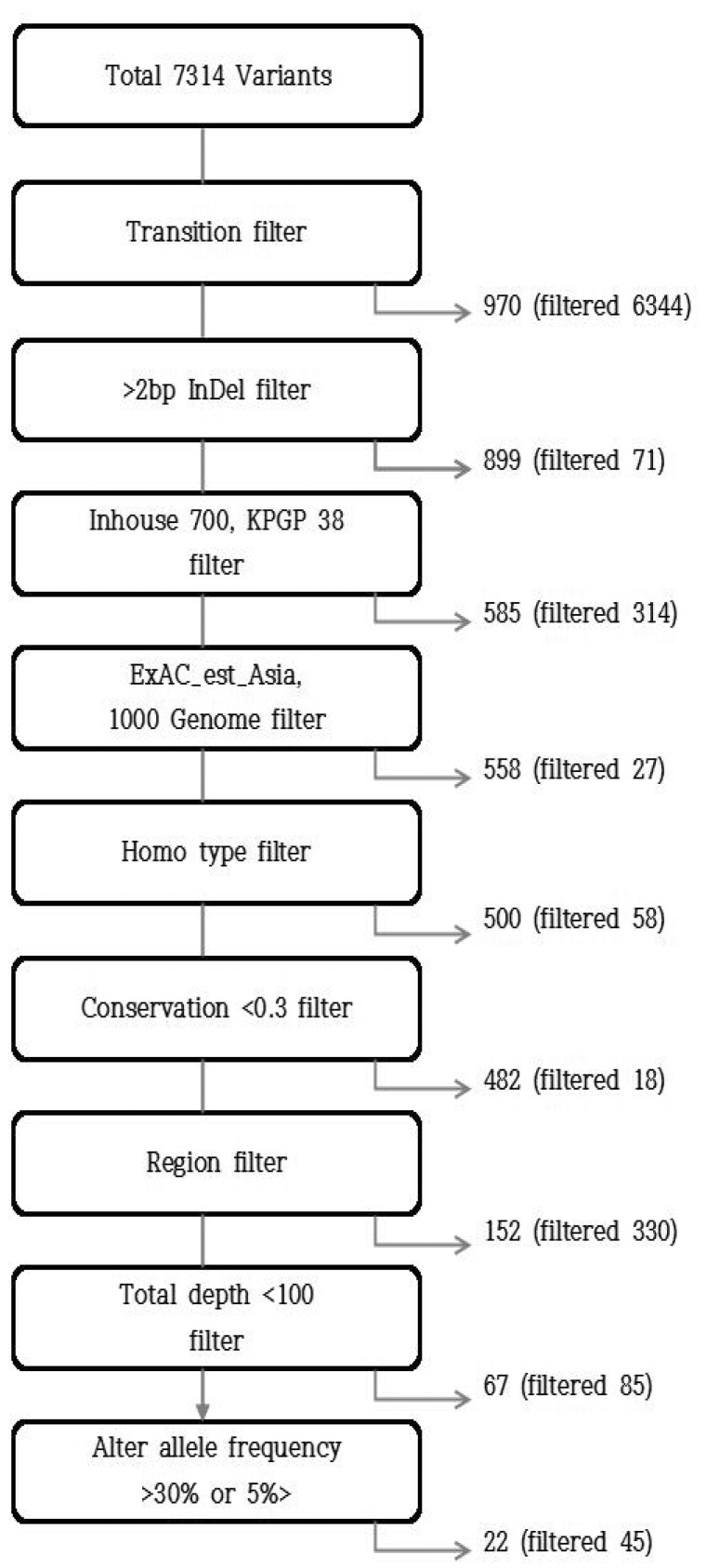
Variant filtering step.
Variant interpretation
To evaluate which mutation could be actionable or those to prioritize, a literature review was done, as was a search for gene function, in some online databases and gene ontology analyses [141516171819].
Sanger sequencing
To validate candidate loci, we conducted PCR amplification and Sanger sequencing. A primer pair for the PCR amplification was designed in the flanking region of each target locus using OligoCalc (http://www.basic.northwestern.edu/biotools/oligocalc.html) and Oligo Analysis Tools (http://www.operon.com/tools/oligo-analysis-tool.aspx). Detailed information on the primers is summarized in Supplementary Table 3. PCR was performed in 20 µL of the reaction mixture, including 10 µL of 2× EF-Taq Pre mix4 (Biofact, Daejeon, Korea), 10 µM of oligonucleotide primers, 1 µL of template, and nuclease-free water. PCR was carried out as follows: first denaturation step of 3 min at 95℃, followed by 25 cycles of 30 s at 95℃, annealing of 30 s at optimal temperature, extension of 40 s to 1 min depending on PCR product size at 72℃, and a final extension for 2 min at 72℃. PCR products were confirmed by gel electrophoresis and purified with a PCR purification kit (Favorgen Biotech Corp., Pingtung Country, Taiwan). Products were sequenced using an ABI3500 genetic analyzer (Thermo Fisher Scientific, Pittsburgh, PA, USA). Sequencing data were aligned with exome sequencing data by the Bioedit program.
Results
Sequencing data and mapping statistics
Supplementary Table 4 summarizes the sequencing data and mapping statistics. The length of the target regions (base pairs, bp) corresponding to the 409 genes was 1,688,650 bp. The total number of mapped reads was 23,172,100, and the average depth of coverage was 1,432×.
Variant types, regions, and single-nucleotide polymorphism (SNP) variant forms of each sample are shown in Supplementary Table 5 and Supplementary Figs. 4 and 5, respectively. A total of 7,314 variants were detected in the five samples, and the number of variants identified in each sample ranged from 881 to 4,099. As shown in Supplementary Fig. 5, most of the SNPs were transition-type (6,344 out of a total of 7,314 variants).
As a result of the filtering steps described above, 22 variants in 25 loci were detected (Table 1). All cases showed one (case No. 3) to 14 variants (case No. 2). The lysine [K]-specific methyltransferase 2D (KMT2D) gene showed the identical SNP variant (missense mutation) in four samples (cases Nos. 2, 3, 4, and 5). Two different variants for the MYH9 gene were identified in one sample (case No. 2). Most of the variants were missense mutations, except for two stop gained variants for ATR and MBD1 and one frameshift variant for FLT4.

Final 22 variants
Variant interpretation
The genes were classified into two groups: likely actionable and unknown significance. The likely actionable group was confined to the genes with a mutation in NKTCL, which has previously and recurrently been identified. Three genes were classified as likely actionable: KMT2D, AT-rich interactive domain-containing protein 1A (ARID1A), and TP53. The other group was classified as the unknown significance groupivarious functions can be present, with molecular function potentially implied in cancer development. The variant interpretation results are summarized in Table 2. Based on their functional categories, the transcription regulation-related gene group, which included four genes related to chromatin modification, was the most common gene group (eight genes). This was followed by a group comprising five genes related to transcription factor/signal transduction.

Variant interpretation results
Sanger sequencing
To confirm the 25 loci, we tried to amplify them using PCR analysis. However, only six loci were successfully amplified in two samples (cases Nos. 1 and 5), due to severe DNA damage in the FFPE samples (Supplementary Fig. 3). We conducted Sanger sequencing of the PCR products. Only one out of the six loci was authentic; one sample (case No. 5) contained a missense mutation (c.3592A>T, p.M1198L) in the BCR gene (Supplementary Fig. 6).
Discussion
To identify somatic mutations of NKTCL, this study used NGS technology, which revealed 25 mutations from 21 genes in five cases. Three genes were classified as the likely actionable group: KMT2D, ARID1A, and TP53. In particular, KMT2D, also known as MLL2, showed the same variant in four cases. Somatic mutations of KMT2D, ARID1A, and TP53 in NKTCL were recurrently reported in recent studies using NGS [202122]. In these studies, KMT2D was one of the most frequently mutated genes, together with STAT3, BCOR, DDX3X, TP53, and ARID1A. These genes were suggested as novel driver genes in the development of NKTCL. We could not identify the mutational status of STAT3, BCOR, or DDX3X, due to the absence of these genes from our cancer panel list.
The KMT2D gene encodes a histone methyltransferase, is classified in the chromatin modification group, and plays an important role in regulating gene transcription. Somatic mutations in the KMT2D gene have been identified in certain cancers [2324]. Most of these mutations result in an abnormally short, nonfunctional histone methyltransferase that cannot perform its role as a tumor suppressor, resulting in the development of cancer. In one of the reports described above, a loss-of-function mutation in KMT2D was identified by RNA sequencing. The result supports the idea that KMT2D may be a tumor suppressor gene in the development of NKTCL and could be a potential therapeutic target. Mutations in this gene were also found in other lymphomas, including relapsed diffuse large B-cell lymphoma, mantle cell lymphoma, and follicular lymphoma [252627]. In follicular lymphoma, an integrated genomic analysis identified early driver mutations in the KMT2D gene, together with other chromatin regulator genes.
Mutations in the ARID1A gene have been found in many types of cancers, including ovarian carcinoma, gastric carcinoma, mantle cell lymphoma, and pediatric Burkitt lymphoma [24262829]. We identified a mutation in ARID1A in one out of the five cases. The ARID1A gene encodes a protein that forms one piece (subunit) of several different SWI/SNF protein complexes. SWI/SNF complexes regulate gene activity (expression) by a process known as chromatin remodeling. The mechanism by which mutations in the ARID1A gene contribute to cancer is unknown, although it is thought that changes in SWI/SNF complexes are involved. These changes may disrupt the regulation of genes that help control the growth and division of cells, which lead to cancer [30].
TP53 is a well-known tumor suppressor gene. The encoded protein responds to diverse cellular stresses to regulate the expression of target genes, thereby inducing cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. We found a mutation in TP53 in one out of the five cases. Mutations in TP53 have been described in NKTCL at various proportions, suggesting some racial, environmental, or lifestyle differences as a possible cause of tumorigenesis, and appear to correlate with more advanced-stage disease [313233]. In other lymphoma studies using NGS, including pediatric Burkitt lymphoma and mantle cell lymphoma, mutations in TP53 were also frequently found [2629].
In summary, the likely actionable group, consisting of KMT2D and ARID1A, in addition to TP53, seems to contain frequently mutated tumor suppressor genes in the development of NKTCL, considering their function and the previous literature. It is likely that other genetic changes, in addition to mutations in these genes, are necessary for the development of this disease.
The other genes (unknown significance group) showed a varied functional spectrum. Like the likely actionable group, most of these genes may have a role as tumor suppressors, considering their molecular function: chromatin modification (SETD2 and KAT6B), transcriptional regulator (MBD1, BCL11B, ZNF521, and PER1), regulation of cytokinesis and structural integrity (CDH11, MYH9, and SYNE1), and DNA damage checkpoint (ATR). Additional RNA sequencing could reveal the functional status (gene expression status) of these genes.
In contrast to the genes described above, most of the remaining genes might be related to activating mutations. The majority of these genes is related to transcription factors and signal transduction (ERG, IKBKB, FGFR1, BCR, and MYH11). Other genes are related to angiogenesis (FLT4 and THBS1) and RNA metabolism (SBDS). In particular, ERG (case No. 1), MYH11 (case No. 2), and BCR (case No. 3) showed the highest allele frequency in each case, and BCR was the only gene that showed a missense mutation result in Sanger sequencing. But until now, genetic alterations of these three genes in NKTCL have not been described, and it is difficult to infer their molecular function or impact in NKTCL, considering their known function up to now.
As described above, this study did not complete Sanger sequencing validation due to severe DNA degradation of the FFPE samples. Although Sanger sequencing is still considered the standard technology for the validation of NGS, it has several limitations. It is insensitive to alterations that occur at an allele frequency lower than approximately 20% [34]. In cancer research, histological variables, such as tumor purity, tumor heterogeneity or subclonality, and the ploidy of the tumor cells, within a sample could significantly affect the sensitivity and specificity of DNA sequencing [35]. However, targeted sequencing with very high sequence coverage, like in this study, allows for the detection of lower-abundance (low allele frequency) somatic variants and thus reveals clinically useful information from stromally contaminated or heterogeneous tumor samples [36]. In this study, the allele frequency of the variants ranged from 5.0% to 27.1%, and only three variants showed an allele frequency of over 20%. As a result, Sanger sequencing was not appropriate for the validation of the mutations in this study.
Because this study involved only five cases, it is difficult to define their mutational spectrum. But, the majority of the genes could be grouped as tumor suppressor genes. Previous NKTCL studies have reported some activating mutations [37383940] and deregulated gene expression in various oncogenic pathways [414243]. This study revealed no mutations in oncogenes that have been reported in previous mutation studies or gene expression studies in NKTCL. However, this study result may imply that the mutations of some tumor suppressor genes and genes involved in transcriptional regulation, transcription factors, and signal transduction may have a role as a trigger, resulting in additional oncogenic events in some pathways and oncogenes in the development of NKTCL and disease manifestation. We suggest that these candidate genes may be used as potential bio-markers for this disease in the future. Further studies of transcriptomic alterations and functional analyses could help identify the molecular function, nature, and clinical significance of these candidate genes in NKTCL and could accelerate the attainment of the goal of personalized medicine based on the genetic alterations in each patient.
References
Supplementary materials
Supplementary data including five tables and six figures can be found with this article online at http://www.genominfo.org/src/sm/gni-14-78-s001.pdf.
Supplementary Table 1
Clinical information
Supplementary Table 2
Sample quality control results
Supplementary Table 3
Primer pairs used in Sanger sequencing
Supplementary Table 4
Statistics of sequencing data
Supplementary Table 5
Variant regions of each sample
Supplementary Fig. 1
Ion AmpliSeq Comprehensive Cancer Panel target gene list.
Supplementary Fig. 2
Representative histological findings and immunohistochemical stains. EBV ISH, Epstein-Barr virus-encoded small RNA in situ hybridization.
Supplementary Fig. 3.
Sample quality control results.
Supplementary Fig. 4
The number of variant types in each sample. SNP, single-nucleotide polymorphism; MNP, multinucleotide polymorphism; INS, insertion; DEL, deletion.
Supplementary Fig. 5
The number of SNP variant forms in each sample. SNP, single-nucleotide polymorphism.
Supplementary Fig. 6
Sanger sequencing results (BCR gene locus).