Ultra-rare Disease and Genomics-Driven Precision Medicine
Article information
Abstract
Since next-generation sequencing (NGS) technique was adopted into clinical practices, revolutionary advances in diagnosing rare genetic diseases have been achieved through translating genomic medicine into precision or personalized management. Indeed, several successful cases of molecular diagnosis and treatment with personalized or targeted therapies of rare genetic diseases have been reported. Still, there are several obstacles to be overcome for wider application of NGS-based precision medicine, including high sequencing cost, incomplete variant sensitivity and accuracy, practical complexities, and a shortage of available treatment options.
Introduction
Next-generation sequencing (NGS) has achieved great advances in medicine and clinical practice, as well as in basic research field. NGS-based precision medicine has mainly focused on cancer [12], given the prevalence, availability of drugs targeting major oncogenic factors, and impact on human health. Another sector of focus through NGS-based precision medicine is ultra-rare diseases.
Ultra-rare diseases can be defined as disorders that occur with a prevalence of below 1 in 2,000,000, although this frequency cutoff can be flexible [3]. Usually, rare disease research is performed with large families for traits with dominant inheritance [45] or patient-parent trios for spontaneous cases [6]. Discovery of causative genetic variants using whole exome sequencing (WES) or whole genome sequencing (WGS) should be followed by in vitro or in vivo model functional studies to fully explain the molecular basis of the diseases and confirm the functionality of the variants [4567].
It is presumed that a large portion of ultra-rare diseases are caused by de novo mutations occurring at functionally important protein coding regions (Fig. 1) [8 9]. Every human zygote is expected to have approximately 70–80 de novo mutations in its genome, and this poses a small chance to have ultra-rare diseases, depending on the position where the de novo mutations lie [810].
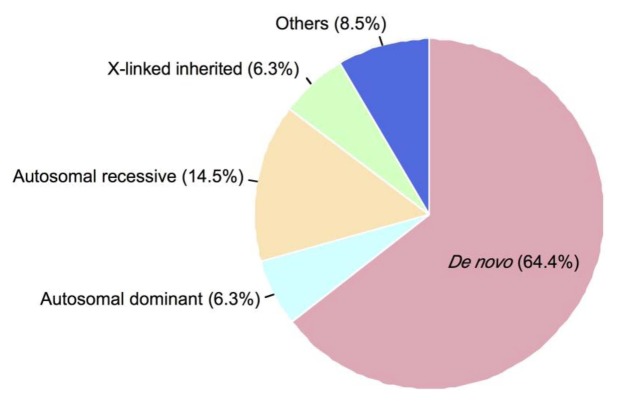
Since NGS was adopted into medical genetics, the number of rare disease-causing gene discoveries has been increasing rapidly, and the genetics of many idiopathic diseases have been elucidated [111213]. In this review, we present the perspectives gained from recent studies on ultra-rare diseases diagnosed by NGS and successful treatments and discuss the potential obstacles for wider application of NGS-based precision medicine.
Cystic Fibrosis
Although cystic fibrosis is not an ultra-rare disease by definition, it presents a good example of precision medicine. Typically inherited in an autosomal recessive manner, patients carry two non-functional alleles of cystic fibrosis transmembrane conductance regulator (CFTR). Recently, an NGS-based diagnostic assay to detect mutations in CFTR was authorized for marketing by the United States Food and Drug Administration (FDA) [14]. In addition, ivacaftor (Kalydeco, Vertex Pharmaceuticals, Cambridge, MA, USA), a potentiator of CFTR, and lumacaftor (Orkambi, Vertex Pharmaceuticals; combination drug with ivacaftor), a corrector of CFTR, were approved by the FDA for the treatment of cystic fibrosis patients with nonsynonymous mutations (http://www.kalydeco.com) and the F508del mutation (http://www.orkambi.com), respectively [15]. NGS-based assays have made it possible to detect certain mutations that can be successfully treated by mutation-specific drugs (Table 1) [16].
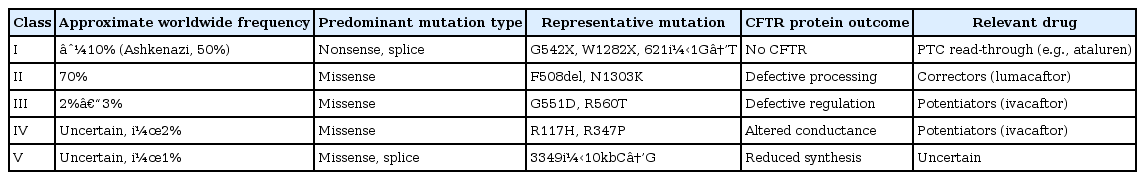
Classes of CFTR mutations
Clinical Practices with NGS-Based Precision Medicine
The famous case of Nick Volker exemplifies the beneficial effect of NGS techniques on patients with atypical and undiagnosed disorders [17]. The cause of his fatal bowel inflammation and colocutaneous fistula turned out to be a mutation in the X-linked inhibitor of apoptosis protein (XIAP) gene by WES, which has important roles in inflammatory signaling and immunity. His disease was almost cured by cord blood transplantation. Following this, a few more cases of precision medicine supported by NGS have been reported. WGS of 14-year-old twins with dopa (3,4-dihydroxyphenylalanine)-responsive dystonia revealed compound heterozygous mutations in the sepiapterin reductase (SPR) gene, encoding sepiapterin reductase. Disruption of the enzyme caused a decrease in the neurotransmitters dopamine and serotonin, and administration of L-dopa with 5-hydroxytryptophan, a serotonin precursor, led to clinical improvements in both twins [18]. Another case was a cancer: the serine/threonine protein kinase B-raf (BRAF) V600E mutation in metastatic rhabdoid meningioma was found by targeted NGS in a 6-year-old girl [19]. She was successfully treated with dabrafenib, a BRAF inhibitor.
Most recently, a more dramatic case was of a 14-year-old girl who had suffered from autoimmune enteropathy since she was 3 months old [6]. A novel de novo mutation in the ligand binding domain of cytotoxic T lymphocyte antigen 4 (CTLA-4) was discovered by WES (Fig. 2) [62021]. The mutation was presumed to be causative after several in vitro immunological experiments. Abatacept, an FDA-approved fusion protein of the extracellular domain of CTLA-4 and the Fc region of immunoglobulin for rheumatoid arthritis, was administered to the patient and restored regulatory T cell function. Clinically, treatment with abatacept without the need for other immunosuppressants reduced diarrhea output by about 67% (from 3 L/day to 1 L/day). Markedly, blood transfusion was not necessary anymore, allowing the patient to be discharged from a 3-year-long hospitalization, merely needing partial parenteral nutrition.
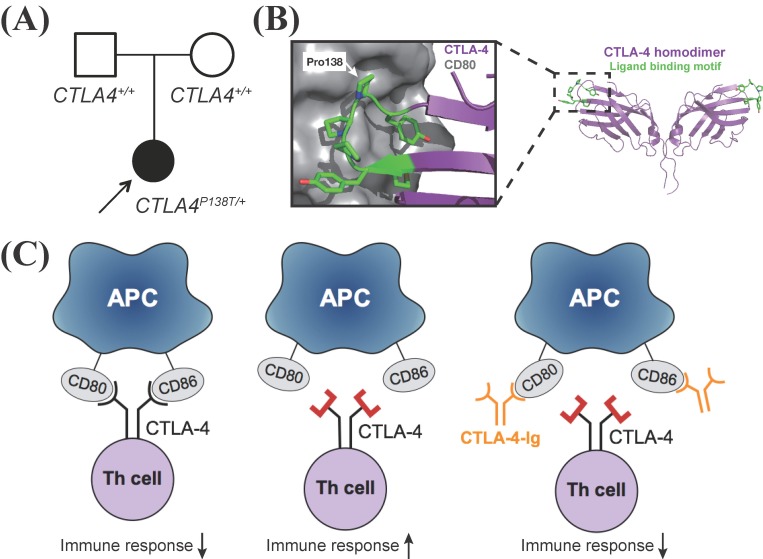
De novo mutation in ligand binding motif of cytotoxic T lymphocyte antigen 4 (CTLA-4). Modified from Lee et al. J Allergy Clin Immunol 2016;137:327-330 [6]. (A) Proband-specific de novo mutation in CTLA-4 shown in the pedigree. (B) Crystal structure of CTLA-4 homodimer (right) (Protein Data Bank [PDB] ID: 3OSK) [20] and close-up view of interaction between CTLA-4 and CD80 (left) (PDB ID: 1I8L) [21]. (C) Models of mutant CTLA-4 failing to bind to CD80/CD86 and regulate immune reactions and rescued by CTLA-4-Ig (abatacept). APC, antigen-presenting cell; Th cell, T helper cell.
Prenatal Diagnosis
Prenatal screening and prediction of ultra-rare disease with NGS are not widely used yet due to their cost and efficacy. But, if the parents of a child with an ultra-rare disease who was diagnosed by NGS plan to have another child, prenatal testing with DNA extracted from chorionic villus sampling or amniocentesis is possible. If the cost of NGS declines, screening of rare genetic diseases with NGS might be routinely performed.
At this moment, a non-invasive prenatal test using NGS is rapidly being introduced to the clinical field, which allows the detection of fetal aneuploidies, such as trisomy 21 (Down syndrome) [22]. This technique uses cell-free fetal DNA (cffDNA) present in maternal circulation and enables non-invasive liquid biopsy, in contrast to invasive procedures, including amniocentesis and chorionic villus sampling. Although it is technically challenging to detect small nucleotide-level mutations from cffDNA at the present time, extensive research in this field has yielded potential in tackling the problem [23].
Obstacles to Precision Medicine and Future Direction
Despite these encouraging cases, there are wide gaps between the diagnosis and treatment of ultra-rare diseases. Targeted and tailored therapies for ultra-rare diseases are usually hard, mainly because of scarce treatment options. To fill these gaps, several conditions should be fulfilled. First, the cost of NGS should decrease further. As of this writing, the cost of WES has fallen to under USD $1,000, which is still a formidable price for a routine application in clinics, while the usage of WGS may still be impractical. Second, the sensitivity and specificity of genetic diagnostics should be improved. Current commercially available exome capture kits covers ~99% of the RefSeq exome, and over 95% of the targeted bases are covered at least eight times with a typical WES run [24], implying that we are achieving fair exome coverage technically. However, the positive diagnostic rate of clinical WES for Mendelian phenotypes remains ~30% [25], suggesting that a substantial portion of the remainder (~70%) might be caused by variants located outside of exons or are not detectable by WES. Although the majority of functionally critical and disease-causing mutations occurs in protein-coding regions, the vast majority of the genome is non-coding and may contain variants with functional significance that has been neglected. Third, processes of NGS data generation should be more simplified. Currently, the typical NGS pipeline consists of library preparation, sequencing, and multiple analysis steps, including read alignments, variant calling, and annotation. For NGS to pervade in clinical practice, the processes from DNA samples to the final results should be more concise. In this context, nanopore sequencing techniques, such as MinION, could function as a good alternative to current NGS technologies if they improve the high error rates and relatively small-scale data output [26]. Fourth, more treatment options, such as targeted drugs and gene therapy strategies, should be provided. The metastatic rhabdoid meningioma with a BRAF mutation and CTLA-4 cases mentioned above are rather exceptional cases, since the genetic alterations were already targeted through commercially available drugs. Nevertheless, the CTLA-4 case is a good example of drug repositioning, which can be an efficient option to accelerate treatments for ultra-rare diseases, given its shorter development time and lower cost [27].
Conclusion
Although they are given less clinical attention and researched less often, ultra-rare diseases have been occurring constantly, because every new baby will have de novo mutations in its genome, posing a small chance of the mutations causing an unseen phenotype. Thus, it would be important to improve prompt and accurate diagnoses and suggest appropriate treatment options for ultra-rare disease patients. Although we still face many technical difficulties to address—such as the high price of NGS, imperfect NGS technology, practical complexities, and a shortage of treatment options—the several cases illustrated here demonstrate successful application of NGS-based precision medicine for ultra-rare diseases, benefiting patient care in the long term. In the near future, NGS will be an essential component in precision medicine regarding its high throughput and relatively low and further decreasing cost and the importance of individual genomic information for personalized strategies.
Acknowledgments
This work was supported by a Research Resettlement Fund for new faculty of Seoul National University.