Non-negligible Occurrence of Errors in Gender Description in Public Data Sets
Article information
Abstract
Due to advances in omics technologies, numerous genome-wide studies on human samples have been published, and most of the omics data with the associated clinical information are available in public repositories, such as Gene Expression Omnibus and ArrayExpress. While analyzing several public datasets, we observed that errors in gender information occur quite often in public datasets. When we analyzed the gender description and the methylation patterns of gender-specific probes (glucose-6-phosphate dehydrogenase [G6PD], ephrin-B1 [EFNB1], and testis specific protein, Y-linked 2 [TSPY2]) in 5,611 samples produced using Infinium 450K HumanMethylation arrays, we found that 19 samples from 7 datasets were erroneously described. We also analyzed 1,819 samples produced using the Affymetrix U133Plus2 array using several gender-specific genes (X (inactive)-specific transcript [XIST], eukaryotic translation initiation factor 1A, Y-linked [EIF1AY], and DEAD [Asp-Glu-Ala-Asp] box polypeptide 3, Y-linked [DDDX3Y]) and found that 40 samples from 3 datasets were erroneously described. We suggest that the users of public datasets should not expect that the data are error-free and, whenever possible, that they should check the consistency of the data.
Introduction
The completion of the human genome project has accelerated the development of many omics technologies that have been used extensively for the genomewide profiling of human samples [1]. Public repositories, such as Gene Expression Omnibus (GEO) and ArrayExpress, now hold data on hundreds of thousands of samples profiled by diverse technologies [23]. For many of the samples in the public repositories, various kinds of clinical information are also included so that interested researchers can use them for their own research interests.
While these public data have enormous potential for research use, it is inevitable that unknown errors may creep into public datasets without being noticed by depositors. For example, Microsoft Excel is notorious for its automatic conversion function, which erroneously changes tens of human gene symbols [4]. But, the errors are not limited to gene symbols and may occur in the clinical information, as well.
Age and gender are some of the most basic clinical information associated with human samples and are most unlikely to be erroneous during data acquisition. While they are basic clinical information, the importance of age and gender in human biology is not trivial. There is strong evidence that men and women differ in terms of development and severity of many common diseases, including cardiovascular diseases, autoimmune diseases, and asthma [5]. Recent clinical studies have revealed an association between several genetic diseases and gender-specific genetic patterns [67].
While analyzing public datasets produced using Infinium 450K HumanMethylation arrays (Illumina Inc., San Diego, CA, USA) and Affymetrix Human Genome U133Plus 2.0 arrays (Affymetrix Inc., Santa Clara, CA, USA), we found many samples that were discordant between clinical gender information and the patterns of gender-specific markers. Importantly, the errors were not limited to a few datasets but were prevalent in many datasets produced by many different laboratories. We advise that the users of public datasets should not expect that these data are error-free and, whenever possible, that they check the consistency of data.
Methods
Data collection and processing
Infinium 450K HumanMethylation array
More than 5,600 samples from 11 datasets were collected from GEO [2]. Before data integration, we calculated the average beta value for measuring methylation levels at each CpG site, ranging from 0 (least methylated) to 1 (most methylated). The individual beta value was dropped if the detection p-value was over 0.05. For all datasets, raw signal intensity files were collected, and from them, methylation level was calculated as β = (max(Cy5, 0)) / (|Cy3| + |Cy5| + 100). A constant of 100 was added to the denominator to regularize β values when both unmethylated and methylated intensities were small.
Selection of gender-specific DNA methylation and gene expression markers
The gender-specific DNA methylation markers of the X chromosome were selected from reported X-linked housekeeping genes [9]. The gender-specific DNA methylation markers of the Y chromosome were estimated using differentially methylated CpG sites in Y chromosomes between males and females (Supplementary Fig. 1). Gender-specific markers were selected based on their beta value distribution in both males and females. As a result, two markers (cg24139739 and cg02869694) were selected as X chromosome markers, and two markers (cg07851521 and cg10835413) were selected as Y chromosome markers. The cutoff values for each marker were cg24139739 < 0.25, cg02869694 < 0.4, cg07851521 > 0.5, and cg10835413 > 0.45 (Supplementary Fig. 1).
The gender-specific gene expression markers were selected from reported gender-specific gene expression patterns in human blood (Supplementary Fig. 2) in a similar way [10]. Two markers (214218_s_at and 224588_at) were selected as X chromosome markers, and two markers (204409_s_at and 205000_at) were selected as Y chromosome markers. The cutoff values for each marker were 214218_s_at < 4.5, 224588_at < 7.5, 204409_s_at > 5, and 205000_at > 5.
Data analysis
Python version 2.7.6 and the Pandas python library version 0.15.2 were used for most data analyses. R version 3.1.0 and ggplot2 version 1.0.0 were used for image production.
Results
Collection of DNA methylation array data of human whole blood with age and gender information
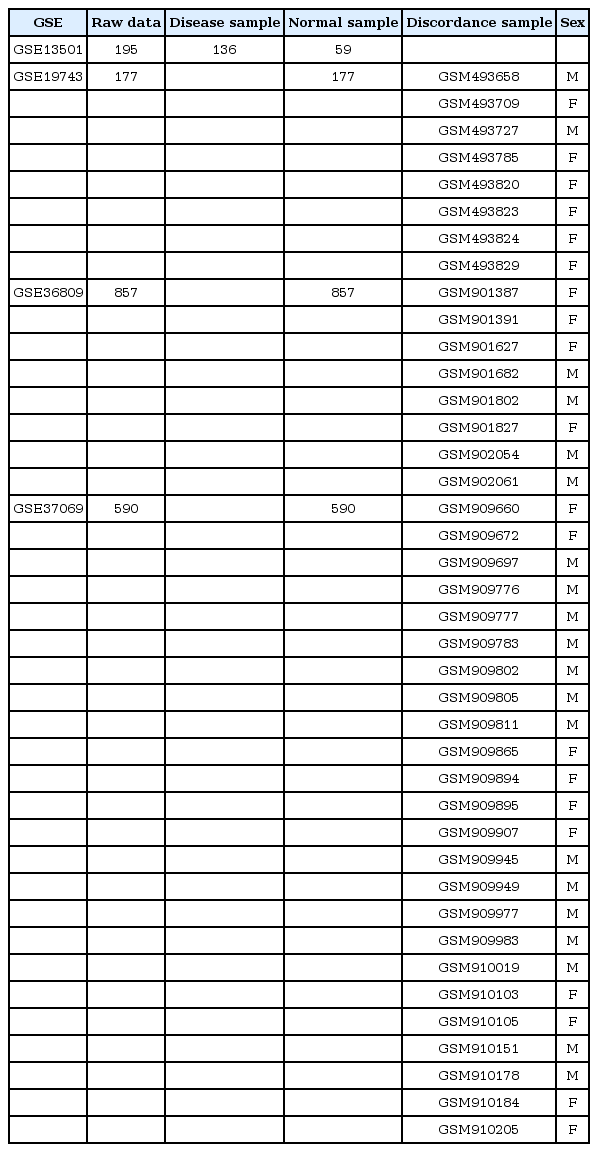
We obtained DNA methylation microarray data from the NCBI GEO database (Fig. 1). We collected datasets of normal human blood samples in which both age and gender data were available. As a result, we collected a total of 4,862 samples for Infinium 450K HumanMethylation array data (Table 1).
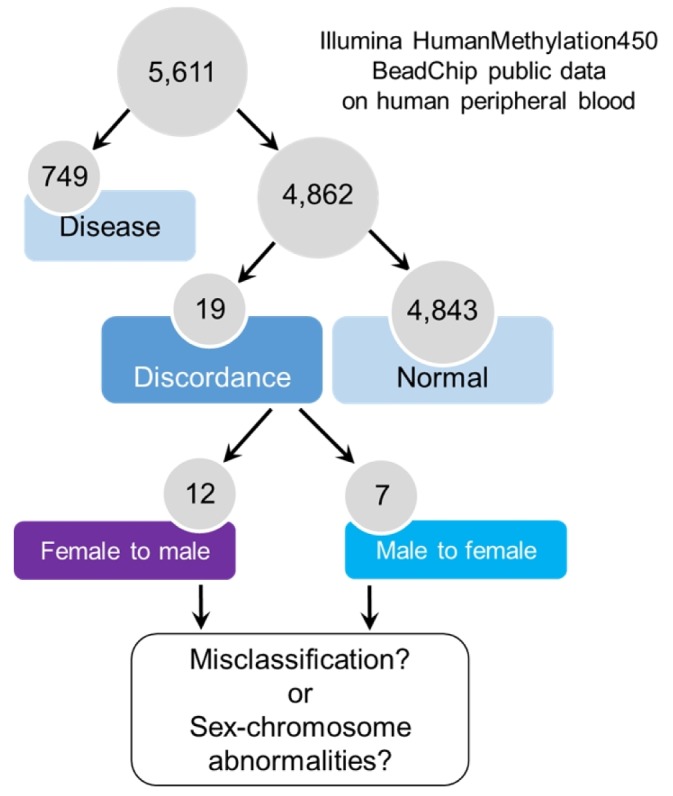
Work flow of the Infinium 450K HumanMethylation array data analysis.

Collected Infinium 450K HumanMethylation array samples and information on discordant samples
Determination of gender by gender-specific markers
The CpG sites of X chromosomes in females are hypermethylated for dosage compensation of female X chromosomes [11]. By analyzing female-specific hypermethylated genes in the X chromosome (Supplementary Table 1), we identified 19 samples (0.39%) in which the methylation patterns of gender-specific CpG markers did not match the given gender information (Fig. 2A). When we analyzed those samples with male-specific hypermethylated genes in the Y chromosome (Supplementary Table 2), we again observed the opposite methylation patterns (Fig. 2B). Importantly, the discordant patterns were observed in eight of 11 datasets from different depositors, suggesting that the errors were not limited to one laboratory (Supplementary Fig. 3).
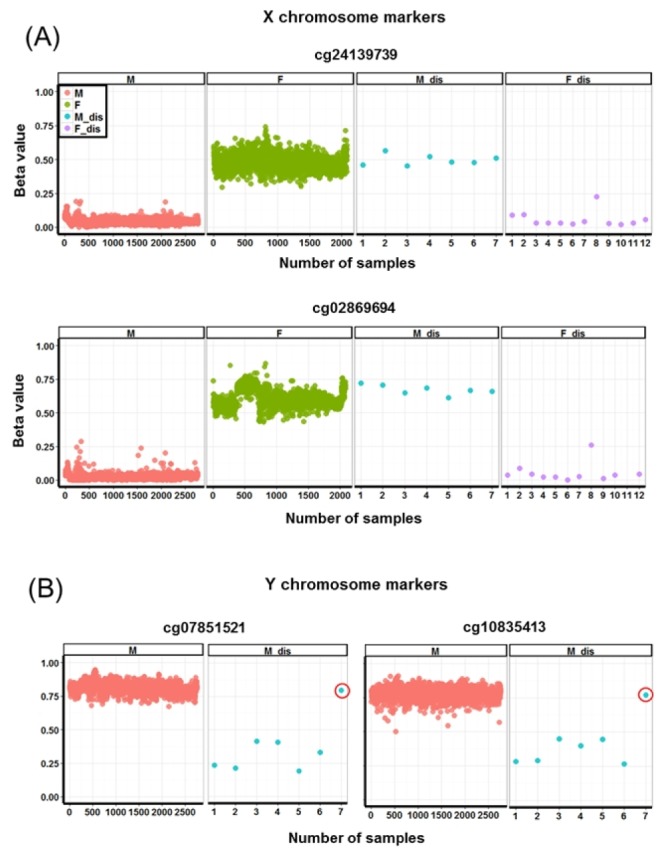
Methylation pattern of gender-specific markers. X (A) and Y (B) chromosome markers. The red circles indicate the discordant male sample showing overlapping distribution of levels for normal male samples. M, male; F, female; M_dis, male samples showing methylation patterns of females; F_dis, female samples showing methylation patterns of males.
Interestingly, we found both types of discordant errors between DNA methylation patterns and given gender information. That is, for some samples, DNA methylation patterns were found to be female-specific while they were designated as males (designated here as discordant-male), and for the other samples, DNA methylation patterns were male-specific while they were designated as female (designated here as discordant-female).
Analysis of markers of chromosome abnormality syndromes
We next analyzed whether the observed discrepancy between methylation patterns of gender-specific genes and the given clinical information could be explained by rare sex chromosome abnormality syndromes, such as Turner and Klinefelter syndromes.
While a normal male inherits an X chromosome and a Y chromosome and a normal female inherits two X chromosomes, several abnormalities in the number of sex chromosomes occur gender-specifically. For females, abnormalities are a result of variations in the number of X chromosomes. For males, abnormalities are due to irregular numbers of the X or Y chromosome or both. The most frequent sex chromosome abnormalities in females are Turner (one X; 1:2,000) and triple X (XXX; 1:1,000), while those in males are Klinefelter (XXY; 1:500) and XYY (1:1,000) syndromes [12131415].
For comparison, we collected Infinium 450K Human-Methylation array data of one Turner syndrome patient and five Klinefelter syndrome patients. The characteristic methylation pattern of Turner syndrome (one X) is the hypomethylation of both X- and Y-specific markers (Fig. 3A). However, we found that most discordant-female samples in our dataset showed hypermethylation patterns in Y-specific markers, suggesting that they had XY chromosomes (a normal male) but not X (Turner syndrome) chromosome (Fig. 3A). Only one discordant-female sample (green circle in Fig. 2A) showed a pattern similar to Turner syndrome. For Klinefelter syndrome (XXY), the expected methylation patterns are the hypermethylation of both male- and female-specific markers. We observed one discordant-male sample in which both male- and female-specific markers were hypermethylated (red circles in Figs. 2B and 3A). However, a close examination of three Klinefelter syndrome-specific markers revealed that the one discordant-male sample did not show the patterns of a Klinefelter syndrome patient (Fig. 3B, Supplementary Fig. 4) [16]. In conclusion, only one sample showed a methylation pattern associated with sex chromosome abnormalities.

Comparison of methylation status between normal and sex chromosome abnormalities. (A) Scatterplot of methylation status of gender-specific methylation markers on X and Y chromosomes. The red circle indicates the sample with suspected Klinefelter syndrome. The green circle indicates the sample with suspected Turner syndrome. (B) Box plot of Klinefelter syndrome-specific methylation markers of normal males, normal females, a sample with suspected Klinefelter syndrome, and Klinefelter syndrome samples. M, male; F, female; M_dis, male samples showing methylation patterns of females; F_dis, female samples showing methylation patterns of males; T, Turner syndrome sample; KS, Klinefelter syndrome sample; KS_sus, Klinefelter syndrome-suspected sample.
Significant difference between clinical age information and predicted age using a DNA methylation age calculator
Recently, several studies have shown that DNA methylation markers can be used to estimate the age of an individual to within 5 y [17]. Because the datasets we analyzed had age information, as well, we tested whether the given age information deviated much from the estimated age from age-specific DNA methylation markers. The age prediction was performed by using the DNA methylation age calculator, a web-based tool that provides a predicted age based on the methylation values of DNA methylation markers of Illumina's Infinium platform [18]. When we compared the absolute age deviation between concordant and discordant samples, we found much larger deviations in age prediction among discordant samples compared with concordant samples (Table 1, Supplementary Fig. 5). This result suggests that the discordant samples are highly likely to have errors both in age and gender information, a scenario that occurs when two samples are mislabeled as each other.
Identification of gender-discordant samples from the analysis of gender-specific gene expression
To see if the same problem occurs in other types of data (e.g., gene expression), we collected gene expression microarray data from the NCBI GEO database using a similar strategy for methylation microarray data (Fig. 4). By applying the same criteria as with the DNA methylation data, we collected a total of 1,683 samples from 4 datasets produced using the Affymetrix U133Plus2 array platform (Table 2).

Work flow of the Affymetrix Human Genome U133Plus 2.0 array analysis.
Collected Affymetrix U133Plus2 array datasets and information on discordant samples
We analyzed the 1,683 samples using several gender-specific genes (X (inactive)-specific transcript [XIST], eukaryotic translation initiation factor 1A, Y-linked [EIF1AY], and DEAD [Asp-Glu-Ala-Asp] box polypeptide 3, Y-linked [DDDX3Y]) [10] and found that 40 samples (2.3%) from 3 datasets were erroneously described (Fig. 5). In the case of Y chromosome markers, some discordant male samples showed overlapping distribution of methylation levels compared with normal male samples. Those samples needed to be compared samples with male sex abnormality syndromes (e.g., Klinefelter syndrome), but unfortunately, we could not find datasets with male sex abnormality syndromes produced using the same platform. However, as these samples showed the opposite pattern in X chromosome markers, we considered them to be putative discordant samples. But, further validation will be needed to confirm these samples.
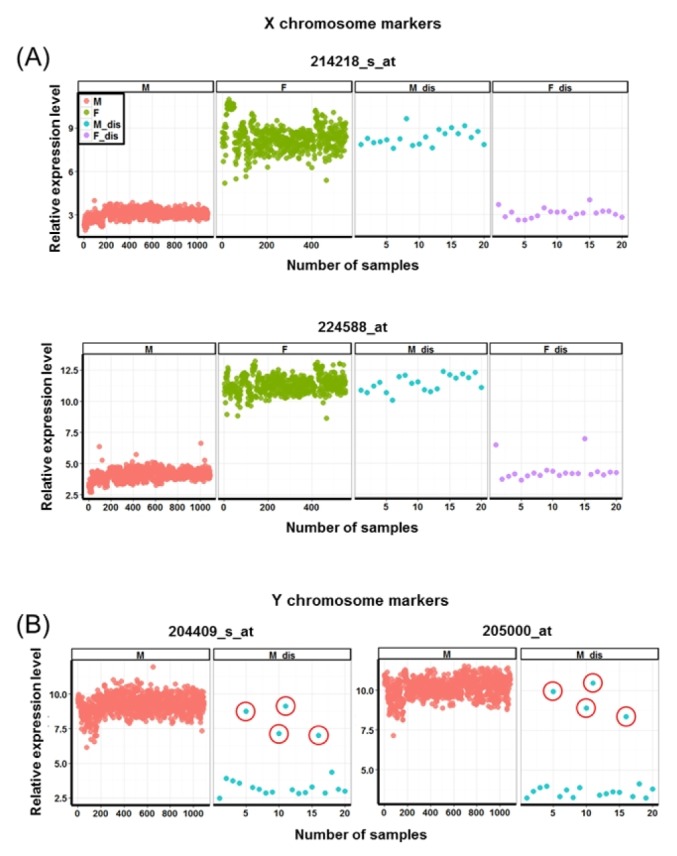
Patterns of gender-specific gene expression of X- and Y-specific genes. X (A) and Y (B) chromosome markers. The red circles indicate the discordant male samples showing overlapping distribution of levels for normal male samples. M, male; F, female; M_dis, male samples showing methylation patterns of females; F_dis, female samples showing methylation patterns of males.
Unlike the previous methylation data, some of the collected gene expression datasets contained technical replicates of the same samples (Table 2). Surprisingly, discordant gender-specific expression patterns were even observed from some of the technically replicated samples (Supplementary Fig. 6).
Discussion
Recently, genomewide omics data have accumulated very fast due to advances in omics technologies (e.g., Illumina's Infinium methylation assay and next-generation sequencing). Now, researchers can exploit public data repositories (e.g., ArrayExpress [3] and GEO [2]) that store data of hundreds of thousands of samples with both genomewide profiling data and associated clinical information. However, as we have shown here, datasets are not error-free, and any type of error can occur in public datasets. Indeed, one paper reported that the aggregate mislabeled blood sample rate was 1.12% at various US institutions in 2013 [19]. Possibly, various kinds of errors (e.g., mislabeling of tubes, mixing of samples) may occur from sample preparation to various steps of the experimental procedures.
When we began the analysis of Infinium 450K Human-Methylation array data, we did not expect errors in gender description to occur so often in several datasets. We thus analyzed if those erroneous samples showed the methylation patterns of gender-specific chromosome abnormality syndromes (e.g., Turner and Klinefelter syndromes) but found that most of the erroneous samples did not. Thus, we concluded that most discordant samples were real human errors. This finding led us to analyze expression datasets of Affymetrix U133Plus2 array data, as well, and again, we found that errors were not rare as well. Fortunately, for transcriptomic and epigenomic datasets, gender-specific markers (Supplementary Tables 1, 2, 3, 4) are well known, so that researchers can check whether the given gender information is concordant with the expected patterns of gender-specific markers. For DNA methylation, a user-friendly website (https://dnamage.genetics.ucla.edu/) [18] is also available for researchers.
In conclusion, we suggest that users of public data should not expect that the data are error-free, and whenever possible, they should check them carefully before use.
Acknowledgments
This work was supported by the Forensic Science Research Project 2014 of the Supreme Prosecutors' Office and a KRIBB research initiative grant.
References
Supplementary materials
Supplementary data including six figures and four tables can be found with this article online at http://www.genominfo.org/src/sm/gni-14-34-s001.pdf.
Supplementary Table 1
A list of the female specific hyper-methylated X chromosome markers
Supplementary Table 2
A list of the male specific hyper-methylated Y chromosome markers
Supplementary Table 3
A list of the female specific expressed genes
Supplementary Table 4
A list of the male specific expressed genes
Supplementary Fig. 1
Box plots of methylation status of normal samples on each gender-specific markers. The red dotted rectang indicate the selected markers. X (A) and Y(B) chromosome markers. M, male; F, female.
Supplementary Fig. 2
Box plots of gene expression status of normal samples on each gender-specific markers. The red dotted rectang indicate the selected markers. X (A) and Y(B) chromosome markers.
Supplementary Fig. 3
Scatter plots of methylation status of the gender-specific markers on each data sets. X (A) and Y (B) chromosome markers. M, male; F, female; M_dis, male samples showing methylation pattern of females; F_dis, female samples showing methylation pattern of males; T, Turner syndrome sample.
Supplementary Fig. 4
Box plots of the Klinefelter syndrome specific methylation markers of the male, female, a Klinefelter syndrome suspected sample and Klinefelter syndrome samples. M, male; F, female; KS_sus, Klinefelter syndrome-suspected sample; KS, Klinefelter syndrome sample.
Supplementary Fig. 5
Comparison of the absolute deviation between chronological and predicted age of the random normal sample (n = 19) and abnormal sample (n = 19).
Supplementary Fig. 6
Scatter plots of gene expression status of the gender-specific markers of technically replicated samples. Each graph represents technically replicated samples.