Compiling Multicopy Single-Stranded DNA Sequences from Bacterial Genome Sequences
Article information
Abstract
A retron is a bacterial retroelement that encodes an RNA gene and a reverse transcriptase (RT). The former, once transcribed, works as a template primer for reverse transcription by the latter. The resulting DNA is covalently linked to the upstream part of the RNA; this chimera is called multicopy single-stranded DNA (msDNA), which is extrachromosomal DNA found in many bacterial species. Based on the conserved features in the eight known msDNA sequences, we developed a detection method and applied it to scan National Center for Biotechnology Information (NCBI) RefSeq bacterial genome sequences. Among 16,844 bacterial sequences possessing a retron-type RT domain, we identified 48 unique types of msDNA. Currently, the biological role of msDNA is not well understood. Our work will be a useful tool in studying the distribution, evolution, and physiological role of msDNA.
Introduction
A retron is a 2-kb-long bacterial retroelement that carries a promoter and three genes: msr, msd, and ret. The msr/msd part of the transcript is processed by a reverse transcriptase encoded by ret, resulting in an RNA/DNA chimera called multicopy single-stranded DNA (msDNA) [1]. The RNA and DNA parts of msDNA are encoded by msr and msd, respectively. The precursor RNA molecule possesses a palindrome between the 5' and 3' ends, forming a doublestranded region between both ends (Fig. 1A). The 5' part of the palindrome is followed by a G residue, which is the branching point for covalent bonding to the cDNA (Fig. 1B) [2]. The msr portion of the precursor RNA possesses two stem-loop structures, which are recognized by the reverse transcriptase [3]. The msd part of the resulting chimera, shown in black in Fig. 1C, is palindromic, also forming a large hairpin structure.

Schematic diagram of multicopy single-stranded DNA synthesis. (A) A precursor RNA molecule with the characteristic palindrome (green). "G" residues in a circle or box (red) are conserved. (B) Reverse transcription yielding cDNA (black). (C) The final msDNA chimera. Adapted from Wikipedia (https://en.wikipedia.org/wiki/Multicopy_single-stranded_DNA).
While msDNA is found in many bacterial species, its biological role is not well understood. Furthermore, msDNA is not well annotated in public sequence databases. In order to facilitate the biological characterization of msDNA, we set out to compile msDNA sequences from known bacterial genomes. Using a few known msDNA sequences, we extracted common sequence and structural features. We screened National Center for Biotechnology Information (NCBI) RefSeq bacterial genome sequences using this rule to compile potential msDNA sequences. Their authenticity as msDNA needs to be confirmed experimentally.
Methods
Prototype msDNA sequences and their features
We used one of the published Escherichia coli msDNA sequences (GenBank accession No. U02551) [2] as the query in a Basic Local Alignment Search Tool (BLASTN) search of NCBI GenBank High Throughput Genomic Sequence (HTGS) division. From the BLASTN hits, we manually curated seven other known msDNA sequences (Table 1). By visual inspection of these prototype msDNA sequences, several common features were recognized: (1) the length of palindrome between the 5' and 3' ends ranged from 5 bp to 10 bp; (2) the distance between both ends ranged from 140 bp to 200 bp; (3) the residues flanking the palindrome were conserved G residues in both strands; and (4) the distance between msr/msd and ret was less than 2 kb.

Prototype msDNA sequences used for the development of detection rules
We developed a local python script that enforces these rules in the candidate retron sequences.
Gene prediction and protein domain search
From the downloaded DNA sequences, genes were predicted with the GeneMark suite [4]. The input sequences were split into 20-kb batches, and GeneMark was run for each batch with the default settings. The predicted genes were translated into protein sequences, which were then scanned for the Clusters of Orthologous Group (COG) domain using Reverse Position Specific (RPS)-BLAST. COG is a microbial domain database distributed by NCBI. RPS-BLAST is one of the BLAST applications distributed by NCBI and uses position-specific scoring matrices (PSSMs) as the target database [5]. We looked for hits of the accession COG3344 (PSSM ID: 225881), which stands for "Retron-type reverse transcriptase (mobilome: prophages, transposons)."
Palindrome motif search
One of the characteristic features of msDNA is a palindrome formed by its 5' and 3' ends. In order to detect such a feature in the potential retron sequences, we used a local installation of the software tool palindrome in the European Molecular Biology Open Software Suite (EMBOSS) bioinformatics software package [6]. It requires several options to be set. The minimum and maximum lengths of a palindrome and the maximum distance between a palindrome pair were set to 5, 10, and 200 bp, respectively, while the number of mismatches between a palindrome pair was set to 1.
Profile search of msDNA
A multiple-sequence alignment of the eight prototype msDNA sequences was generated with ClustalW (Fig. 2). We then built a hidden Markov model (HMM) profile from the multiple-sequence alignment using the software tool HmmerBuild in the HMMER package [7]. The potential retron sequences were screened using HmmerSearch with the HMM profile [7]. The cutoff for a specific hit was determined by leave-one-out crossvalidation as follows: (1) among the eight seed sequences, one was set aside, and the remaining seven were used to build the profile; (2) what was set aside was then evaluated using the profile; (3) this process was repeated for each of the eight seeds, and the minimum score was defined as the cutoff (45.0).

Multiple-sequence alignment of prototype multicopy single-stranded DNA sequences. The msr/msd portion of each genomic DNA sequence in Table 1 was used to construct the alignment with ClustalW. The corresponding secondary structures, predicted with CentroidFold, are shown in bracket notation. The conserved positions are colored with Jalview.
RNA secondary structure prediction
The precursor RNA molecule of msDNA forms a characteristic secondary structure. We used a local installation of the software tool CentroidFold [8] to predict the secondary structure of the msr/msd portion of the candidate retron. As the palindrome terminates with a G residue in both strands, the 5' of which is the branching point for covalent bonding to the cDNA (Fig. 1B), sequences without these G residues were removed. The web service version of RNAfold [9] was also used to visually confirm the final prediction set.
Results
Compilation of candidate retron sequences from known genome sequences
Historically, msDNA has been discovered exclusively in bacteria. A keyword search of NCBI RefSeq bacterial genomes with "reverse transcriptase" returned 34,637 hits, as of September 2, 2015. Following the gene prediction of these genomes using GeneMark, a protein domain search for "Retron-type reverse transcriptase (mobilome: prophages, transposons)" using RPS-BLAST resulted in 16,844 candidate retron sequences.
Alternatively, filtering out nonretron-type reverse transcriptase sequences can be also achieved by extracting protein sequences from the RefSeq annotation, followed by the domain search. Instead, we chose to predict genes from the genome sequences using GeneMark, followed by the domain search. Our approach does not rely on annotation information and has the potential to be applied to newly sequenced genomes as an independent tool.
Automatic filtration of candidate msDNA sequences
The candidate retron sequences identified above were filtered by the presence of palindrome motifs. Using the local python script, we searched the 16,844 bacterial sequences for palindrome motifs 5–10-bp-long and separated by less than 200 bp within 2 kb upstream of ret, yielding 7,428,332 such hits. These hits were then screened by the HMM models that were developed based on the prototype msDNA sequences. There were 3,865 hits surpassing the crossvalidation threshold of 45.0. Using a local installation of CentroidFold, the RNA secondary structures were predicted for the remaining hits, and those without the proper double-strand pairing between the 5' and 3' ends were removed. Furthermore, those without the conserved flanking G residues were also filtered out. The whole process, which was wrapped in a Linux shell script, resulted in 625 hits.
Manual curation and clustering of msDNA sequences
The final set of 625 candidate sequences that were identified by an automatic process was highly redundant. The exact copies were pruned, giving rising to a set of 88 unique sequence types. As shown in Fig. 1A, the true msDNA should possess two hairpins in msr and a long hairpin in the msd region. In order to confirm this complex overall topology of msDNA, we examined the RNA secondary structure plots of the 88 candidates visually. In fact, we removed 40 sequences that did not form the appropriate overall topology. See Fig. 3 for a few representative surviving examples. Among the original 625 sequences, 525 can be mapped to these 48 unique sequence types (Supplementary Table 1).
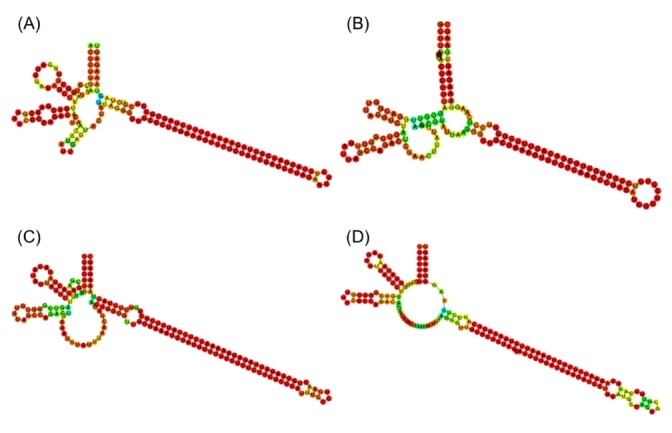
Representative examples of secondary structures of msr/msd precursors of predicted multicopy single-stranded DNA sequences. (A) NZ_AERV01000006.1 Salmonella enterica (J, 86). (B) NZ_ADUL01000064.1 Escherichia coli (B, 3). (C) NZ_AELI01000009.1 Vibrio cholerae (E, 48). (D) NZ_CBWG010000185.1 Escherichia coli (N, 504). The single letters and numbers in parentheses refer to the clustering group and sequence number in Supplementary Table 1.
In order to cluster the remaining 48 sequence types further, allowing small divergences within clusters, multiple-sequence alignment was performed with Multiple Alignment using Fast Fourier Transform (MAFFT) [10]. From the resulting dendrogram, we recognized 20 groups by splitting the tree at a branch length of between 0.12 and 0.20 (Fig. 4). The species distribution of the 525 msDNA sequences over the 20 groups is given in Table 2 and Supplementary Table 1.
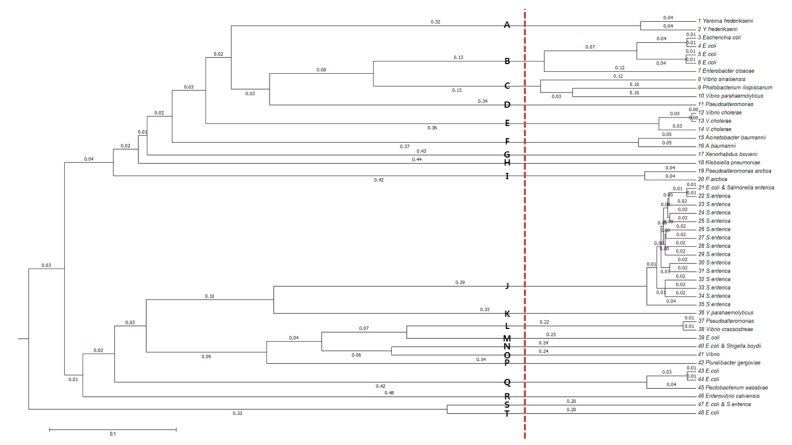
Dendrogram of the final 48 multicopy single-stranded DNA sequence types. The plot was generated with a multiple-sequence alignment program, Multiple Alignment using Fast Fourier Transform (MAFFT). Distinct clusters are marked with symbols, A–T.

Distribution of 525 msDNA sequences in each group
Discussion
We developed a rule-based protocol to detect msDNA in a given genomic DNA sequence. The rule is based on the presence of a 5–10-bp-long palindrome motif in the precursor transcript that is less than 2 kb upstream of a retron-type reverse transcriptase gene. The rule is augmented by enforcing a sequence similarity with known msDNA sequences. This step is implemented with an HMM profile search method. While this was a powerful filtration step, reducing the hits by about 1/20, it would inevitably miss some true msDNA sequences that are distantly related to the prototypes given in Table 1. As more distinct msDNA sequences are discovered, the panel of the seed sequences must be updated.
The list of potential candidate msDNA sequences was filtered by conformation to the known topology of the RNA secondary structure. The RNA secondary structure prediction programs usually output the result in terms of Vienna dot bracket notation [9]. As the manipulation of the string is not straightforward, some manual curation was involved. In the future, we will explore the possibility of automatic implementation, which is critical for web-based service of the prediction tool. To our knowledge, this is the first large-scale annotation of msDNA in all publically available bacterial genome sequences. As our work is computational in nature, the list we compiled can include some false positives. Its authenticity should be validated experimentally. Nevertheless, our list of msDNA sequences and their species distribution profile constitute a useful resource for the future study of msDNA, as its biological role is still elusive.
Acknowledgments
This work is based the Bachelor's degree thesis of WY at Soongsil University, Seoul, Korea. We acknowledge the financial support from the National Research Foundation of Korea (NRF-2012M3A9D1054705 and 2010-0023759).
References
Supplementary materials
Supplementary data including one table can be found with this article online at http://www.genominfo.org/src/sm/gni-14-29-s001.pdf.
Supplementary Table 1
Information of 525 msDNA sequences