Introduction
Infectious diseases are major dreadful threat to human life, spread through many causative agents like bacteria, fungus, virus, etc. Therefore, development of potential drug is highly essential to fight against infectious diseases. Generally, methods of traditional drug design are tedious, time consuming, and cost intensive. Hence, several multidisciplinary approaches have gained research attention to reduce the time and cost during drug development process. In silico drug design is one of such approach, provides remarkable opportunity for identification of novel lead compounds as comparison to other conventional methods. Drug target identification is an important step involved in computer aided drug design process. In this context, the present study is planned to identify a potential drug target against infectious diseases caused by Bordetella petrii using various computational tools and techniques. B. petrii is a gram-negative coccobacilli of the phylum Proteobacteria, has been found exclusively in close association with humans and various warm-blooded animals due to its independent existence as an environmental facultative anaerobe [1]. B. petrii exhibits the host associated properties, and its strains have been detected from environmental samples like microbial consortia degrading aromatic compounds, marine sponges, polluted soil and a grass root consortium [2,3,4,5,6]. The genome of B. petrii contains 5,287,950 base pairs, and has been observed with greatest numbers of expressed virulence factors [7]. Infectious diseases like mandibular osteomyelitis [8], suppurative mastoiditis [9], chronic pulmonary diseases [10], respiratory infection in case of bronchiectasis and cystic fibrosis [11,12] have been reported due to B. petrii infection. Again, exposure to soil or water containing B. petrii may lead to local colonization and eventually serious nosocomial infections [13,14]. Some other bacteria like Escherichia coli [15], Pseudomonas aeruginosa [16], Chlamydophila pneumoniae [17], Staphylococcus aureus [18], and Haemophilus influenzae [19] have also been identified as a cause for such types of infections. Many Food and Drug Administration (FDA) approved drugs have been used against these pathogens, but those drugs have less effect on B. petrii due to development of substantial resistance [10], generated an opportunity to identify a potential drug target as well as to discover a novel lead compound against B. petrii using computational techniques. Again, due to adverse effect of most of the anti-infectious drugs of synthetic origin, natural compounds with anti-infectious properties have been tested in both in vivo and in vitro to be an alternative approach towards the treatment. In this context, some of the natural compounds such as ajoene (Allium sativum), allicin (A. sativum), cinnamaldehyde (Cinnamomum cassia), curcumin (Curcuma longa), gallotannin (active component of green tea and red wine), isoorientin (Anthopterus wardii), isovitexin (A. wardii), neral (Melissa officinalis), and vitexin (A. wardii) have been acknowledged with anti-bacterial properties [20,21,22,23,24,25]. Curcumin is a naturally occurring polyphenolic compound, a common component of Indian spice turmeric, having anti-tumor, anti-oxidant, and anti-inflammatory properties. It has been reported as an agent to reduce bacterial colony formation and inhibit lung cancer progression in high-risk chronic obstructive pulmonary disease (COPD) patients [20]. Similarly, gallotannin is an active component of green tea and red wine has been observed as a potent inhibitor of calcium-activated ClŌĆō channel, may be used for the treatment of cystic fibrosis [21]. Again, in vitro study of three flavone C-glycosides such as isoorientin, vitexin, and isovitexin isolated from A. wardii have been reported as potent inhibitors for interleukin-8, and matrix metalloproteinase-1, an inflammatory marker in case of COPD [22]. Moreover, most of the bacterial infection caused due to formation of bio film in the host. In this regard, an active compound cinnamaldehyde of C. cassia have been identified as a strong inhibitor against quorum sensing (QS) biofilm formation due to E. coli, and Vibrio harveyi [23] infection. Similarly, two extracts of A. sativum namely allicin and ajoene have been accounted for inhibitory effect against P. aeruginosa and E. coli [24]. Likewise, neral is an active component of M. officinalis, has been reported as anti-bacterial, anti-fungal, and anti-oxidant properties against P. aeruginosa, Klebsiella pneumoniae, S. aureus, and Citrobacter koseri infection [25], and may be applied for several infectious diseases including nosocomial infection. As, all of these nine natural compounds have been established with anti-bacterial properties, may be useful for treatment of B. petrii infection, encouraged authors to elucidate their inhibitory effect against the putative drug target of B. petrii through in silico approach.
Methods
Subtractive proteomics
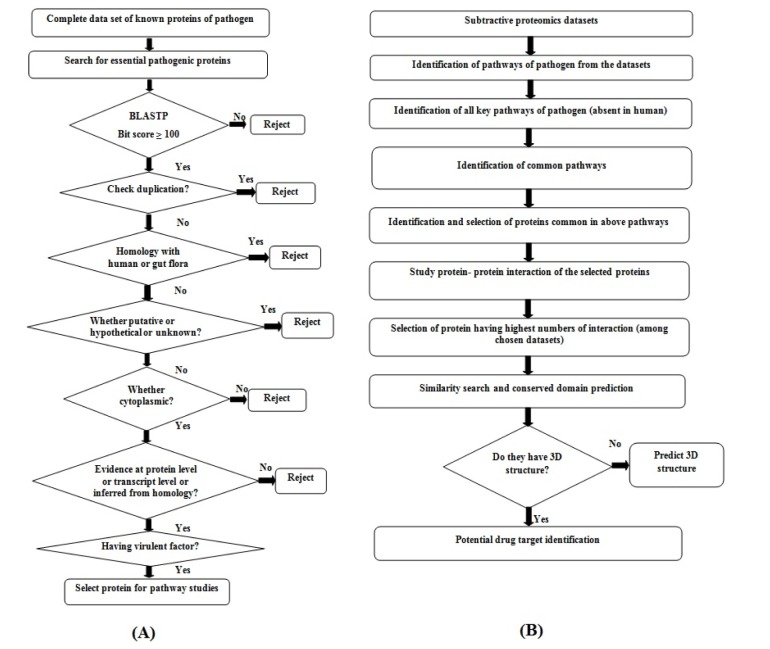
The complete protein datasets of B. petrii (whole genome project accession No. PRJNA28135) were retrieved from Genome database of National Centre for Biotechnology Information (NCBI) web server (http://www.ncbi.nlm.nih. gov/genome/). The essentiality search [26] was carried out through the Database of Essential Gene (DEG) for screening out the essential proteins responsible for the survival of B. petrii in the host. The bit score parameter was considered as greater or equal to 100 for homology search using Basic Local Alignment Search Tool (BLAST) while accessing DEG (http://tubic.tju.edu.cn/deg/blast.php) database, to select bacterial essential proteins. The non-redundancy search was performed through CD-HIT (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi) web server to remove the redundant proteins among selected essential proteins of B. petrii. Further, by considering the resulted proteins of B. petrii, the sequence similarity search was carried out against human [27,28,29] followed by human gut flora microorganisms [30,31] using BLASTP algorithm to eliminate proteins homologous with human and human gut flora microorganisms respectively. During similarity search, the threshold for expected value (e-value) was fixed as greater or equal to 0.001. Furthermore, the protein sequences which are putative, unknown, hypothetical, probable, conserved hypothetical and unnamed protein were removed manually. The study of sub cellular localization was performed to identify the cytoplasm proteins among the selected proteins [32] using CELLO (http://cello.life.nctu.edu.tw/). Generally, proteins are annotated and ranked as per their evidence level in UniProt database which is also a factor to be considered while selecting putative drug targets. Afterwards, the Virulence Factor Database (VFDB) was explored, and the virulence proteins were screened out (http://www.mgc.ac.cn/VFs/blast/blast.html) among the selected targets of B. petrii through VFDB BLASTP search against VFDB core data sets with consideration of threshold e-value as greater than 0.0001 [31]. The above methodology is described through a flow diagram (Fig. 1A).
Pathway analysis
The participation of virulent proteins in any unique pathways of pathogen could be ensured and analyzed through the exploration of databases, is a common procedure to be followed while predicting putative drug targets. In this study, the pathway analysis was performed using Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). Here, the exploration of KEGG database for pathway study was conducted twice. In the first case, the search was carried out for all obtained virulent proteins against existing pathways of B. petrii using KEGG BLAST algorithm. Secondly, unique pathways of B. petrii (pathways present in B. petrii, not in human) were sort listed from the KEGG database by comparing all pathways between B. petrii and human. Further, selection of common unique pathways [26,32,33] was made between the observed pathways of selected targets and obtained unique pathways of B. petrii. Furthermore, the selection pathways which are essential for the survival of the pathogen were made after detail inspection of obtained pathways. Finally, the key proteins regulating those pathways of B. petrii were considered as putative drug targets (Fig. 1B).
Protein-protein network study
The putative drug targets of B. petrii obtained after pathway analysis were subjected for protein-protein network analysis using STRING (http://string-db.org/) web server. Here, the interaction between the drugs targets were quantified on the basis [34] of confidence value (0.007) and the level of interaction (maximum as 50). The drug targets observed as having higher numbers of interaction with other drug targets among the chosen data sets were considered for further analysis (Fig. 1B).
Similarity search and domain prediction
Similarly search was carried out for selected proteins using BLASTP algorithm against Protein Data Bank (PDB) database (http://www.rcsb.org/pdb/). The closest homolog for each selected target protein was considered as template based on lowest e-value and highest percentage of similarity The conserved domain region of potential drug target was predicted using conserved domain database (CDD) of NCBI web server (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) with default parameters (Fig. 1B).
Structure prediction and evaluation
Phyre2 (Protein Homology/Analogy Recognition Engine) server was used for modelling of three dimensional structure of putative drug target of B. petrii. The algorithm of Phyre2 server predict three dimensional structure of a protein sequence using the principles and techniques of homology and/or fold recognition, as structural folding pattern is more conserved in evolution than its amino acid sequence. Hence, a protein of interest (drug target) can be modelled with reasonable accuracy basing on a distantly related protein with known (template) structure [35]. The energetically stable structure was obtained through structural refinement and energy minimization using ModRefiner algorithm [36] of Zhang Lab web server (http://zhanglab.ccmb.med.umich.edu/ModRefiner/). The structural superimposition of refined model with template model was performed using Discovery Studio 3.5 suite. The structural quality and reliability of refined model were evaluated using RAMPAGE [37], ProSA-web [38], Protein Quality Predictor (ProQ) [39], and ERRAT [40] web tools.
Preparation of natural compounds
The chemical structures of nine phytochemicals (ajoene, allicin, cinnamaldehyde, curcumin, gallotannin, isoorientin, isovitexin, neral, and vitexin) were downloaded from PubChem database of NCBI web server (http://ncbi.nlm.nih.gov/pccompound). The correction of structural geometry and energy minimization were carried out using Discovery Studio 3.5 suites.
Molecular docking
Molecular docking between the predicted model of potential drug target with each of the nine natural compounds was conducted using AutoDock-4.2 algorithm (http://autodock.scripps.edu/), by considering the protein structure as rigid whereas ligands were kept as flexible. AutoDock algorithm requires pre calculated grid maps, one for each atom type present in the flexible molecules being docked, and it stores the potential energy arising from the interaction with rigid macro molecules [41]. Ligands were docked around the identified conserved region within the functional domain of the selected drug target. The favorable poses were selected on the basis of lowest binding energy calculated by AutoDock which indicates for most favorable binding conformation between receptor and ligands. Docking procedure was carried out as per normal methodology for protein-ligand docking [42,43]. The visualization and analysis of interaction between docked complexes were made using Discovery Studio 3.5 suites.
Results and Discussion
Screening of putative drug targets
Availability of whole genome information of B. petrii DSM 12804 (accession No. PRJNA28135) in Genome database of NCBI web server encouraged authors to retrieve the entire set of 5,031 protein sequences in order to identify a potential drug target against it. Primarily, screening was made through identification of essential genes of B. petrii, considered as primitive strategy [26] for target identification. Here, out of total 5,031 proteins, only 1,841 were obtained as essential proteins and hence suspected as primary targets of B. petrii. To remove redundancy [29,30] among the putative drug targets, a non-redundancy search was carried out which resulted 1,802 proteins as non-redundant. Search for sequence homology with human proteins is an essential step to be followed for identification of proteins present only in pathogen but not in human [27,28,29]. Again, due to symbiotic relationship among human and human gut micro biota, homology search between proteins of pathogen and human gut flora microorganisms is also a crucial step [30,31] to be performed to eliminate the target proteins which are homologous to human gut flora microorganism. The resulted 799 numbers of proteins of B. petrii were not shown any significant similarity with human proteome as well as human gut flora proteome, hence were selected for downstream analysis. Furthermore, the reduction in protein dataset was done by selecting proteins reported as active and as well as functional. Out of 799 proteins, only 509 numbers of functionally characterized proteins were selected using the information available at UniProt database. As suggested by the literature, only cytoplasm proteins are suitable drug targets whereas membrane bound proteins are generally considered for vaccine development [32]. Among them only cytoplasm proteins (334 numbers) were considered for further study. Furthermore, screening was carried out on the basis of evidence report. As of UniProt database report, the evidence which supports the existence of proteins are classified as: (1) experimental evidence at protein level; (2) experimental evidence at transcript level; (3) protein inferred from homology; (4) protein predicted; and (5) protein uncertain. Out of these evidences, evidence at protein level is the best choice for protein existence, as it indicates the existence of the protein experimentally. But in the current investigation, not a single protein has been observed satisfying this criterion. Only 135 proteins which were inferred from homology subjected to virtual study. Identification of pathogenic proteins having virulence factor is also obligatory [31] while screening a drug target against any pathogenic microorganism. Therefore, homology search was carried out between 135 numbers of proteins and the core virulence proteins of B. petrii using VFDB BLAST. The result suggested, out of 135 selected proteins only 26 number of proteins found with virulence factor, which strongly supported for further study to predict potential drug target against B. petrii (Figs. 1 and 2).
Analysis of metabolic pathways
The pathway analysis was conducted to screen out the putative drug targets and their involvement in important and unique pathways of the pathogen B. petrii which are essential for the survival of the pathogen. The preliminary search in KEGG database suggested for involvement of 22 proteins in different important pathways of B. petrii, and therefore the dataset of putative drug target reduced from 26 to 22. Further, exploration of all available pathways for both human and pathogen was conducted and compared, resulted 38 unique pathways present in B. petrii but absent in human. Furthermore, the comparison was made between the identified important pathways of B. petrii for 22 drug targets and 38 unique pathways identified in B. petrii, resulted 12 putative drug targets (Table 1) contributing towards the 3 common and unique pathways of B. petrii namely, lipopolysaccharide biosynthesis, two component system, and bacterial chemotaxis. As per literature suggest, lipopolysaccharide biosynthesis is an essential pathway [44] for the survival of B. petrii, a major component of the outer membrane of gram negative bacteria and referred as endotoxin. It is comprised of a hydrophilic polysaccharide and a hydrophobic component referred as lipid A which is responsible for major bio reactivity of endotoxin [44], also serves as a physical barrier providing protection towards bacteria from its surroundings [45]. Intense inspection on lipopolysaccharide biosynthesis pathway (Fig. 3) suggested, out of 12 selected putative drug targets (Table 1) only eight numbers (lpxH, kdsB, lpxK, lpxD, lpxB, kdsA, lpxC, and Bpet0453) were as key regulatory proteins [46]. Again, involvement of two components namely, amino acid and sugar metabolism and pentose phosphate pathway were found as regulating the complete pathway. lpxC, lpxD, lpxH, lpxB, and lpxK proteins are involved in amino acid and sugar metabolism, and among them lpxH was discarded as the synthesis of lpxB could alternatively possible by lpxD. Further, lpxB and lpxK were also rejected as potential drug target as the regulation of kdtA and waaA through lpxB and lpxK were otherwise possible by the proteins involved in pentose phosphate pathway. Furthermore, Bpet0453 (5.3.1.13), kdsA (2.5.1.55), and kdsB (2.7.7.38) proteins were found as key proteins of pentose phosphate metabolism components (Fig. 3), and regulating synthesis of kdtA and waaA, but the synthesis of same proteins was also control by proteins involved in amino acid and sugar metabolism to carried out the whole lipopolysaccharide biosynthesis. So the key proteins kdsA, kdsB, and Bpet0453 of pentose phosphate pathway were discarded. Finally, two proteins namely lpxD and lpxC were selected on the basis of their highly involvement in the regulation of lipopolysaccharide biosynthesis pathway than any other proteins (Figs. 2B and 3).
Protein network analysis
Protein network analysis is generally used for selection of drug targets having interaction with maximum numbers of other drug targets, an efficient technique to discover a potent drug target against pathogens [34]. Here, the biological interaction was studied for 2 obtained drug targets of B. petrii, and the assessment supported for both lpxD (with lpxC, lpxB, kdsA, kdsB, lpxH, lpxK, and bpet0453) and lpxC (with lpxD, lpxB, kdsA, kdsB, lpxK, and bpet0453) proteins showing interaction with other drug targets among the chosen data set (Table 2, Fig. 4). Therefore, lpxD and lpxC proteins were strongly recommended to be potential drug targets against the pathogen B. petrii.
Identification of potent drug target and domain prediction
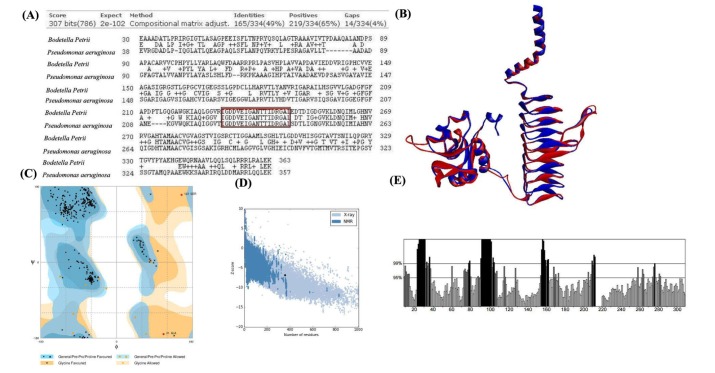
Further, template structure identification for lpxD and lpxC proteins were undergone through similarity search against PDB. The template protein structure (PDB ID: 3PMO) from P. aeruginosa was selected for the drug target lpxD of B. petrii with 91% query coverage and 49% of identity where as template structure (PDB ID: 4FW3) from P. aeruginosa for lpxC protein of B. petrii with 88% query coverage and 56% of identity (Table 3). Though, it has been already established that lpxC catalyzes initially during the biosynthesis of the lipid A component of lipopolysaccharide and may be a potent drug target to block the lipopolysaccharide pathway of pathogens [47], the current investigation suggested, the status of lpxC protein B. petrii as un reviewed (Table 3) and have shown less number of interaction with other drug targets in comparison to lpxD (Table 2). Again, as per literature report, lpxD has a key role in biosynthesis of the lipid A component of lipopolysaccharide, and its inhibitors are also potentially attractive as antibacterial agents, as there is no close similarity found with human proteins [48]. Also, lpxD has a vital role in bio film formation which is causing nosocomial infection [16] and the status obtained from UniProt web server as reviewed (Table 3). This seems lpxD could be a potential drug target against B. petrii infection. Therefore, lpxD was proposed as a potent drug target subjected for further in silico study. Through BLAST search and inspection in CDD database, the conserved amino acid resides within the left-handed parallel beta helix (LbH) domain of lpxD were identified (Fig. 5A), containing three imperfect tandem repeats of a hexapeptide repeat motif (X-[STAV]-X-[LIV]-[GAED]-X).
Structure prediction and refinement of proposed drug target
Due to absence of experimental structure the tertiary model of lpxD (UniProt ID: A9INS9) protein was predicted by the single highest scoring template i.e., lpxD protein of P. aeruginosa (PDB ID: 3PMO, chain A) using phyre2 [35] web server with 100% confidence. Protein attains the lowest free energy during the native state; therefore the stability of the predicted model was improved through energy minimization [36] procedure, supported by the Root Mean Square Deviation score 0.612 obtained through the structural superimposition of lpxD models both before and after energy minimization (Fig. 5B). The refined lpxD protein model is publically available in Protein Model Database with PMDB ID (PM0080538).
Model evaluation of proposed drug target
The correctness and quality of predicted lpxD protein model of B. petrii was verified through RAMPAGE [37], ProSA-web [38], ProQ [39], and ERRAT [40] online server. The RAMPAGE analysis revealed 99.5% residues were folded properly suggesting good backbone folding pattern (Fig. 5C). Further, Z-score for lpxD model calculated by ProSA-web to be ŌĆō6.84 (Fig. 5D) which is well within the range of native conformation of experimental structures. Furthermore, ProQ computed a Levitt-Gerstein (LG) score of 2.785 and Max sub score to be 0.225 for the predicted model. A ProQ LG score >2.5 is necessary for suggesting a model is of good quality [39], implying high accuracy level of predicted structure. Also, the overall quality factor for lpxD model of B. petrii was predicted as 73.23% (Fig. 5E) by ERRAT web server. All of these evaluations recommended the reliability of the predicted lpxD model of B. petrii.
Selection of natural compounds
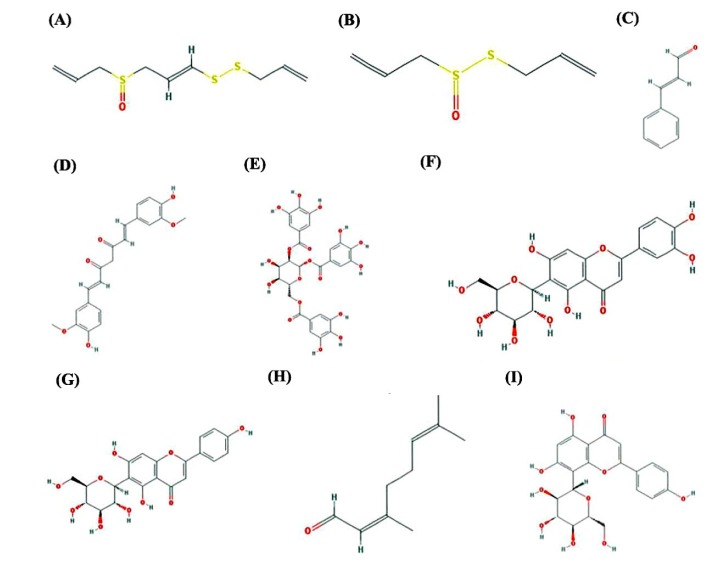
As per literature survey, nine natural compounds such as ajoene (A. sativum), allicin (A. sativum), cinnamaldehyde (C. cassia), curcumin (C. longa), gallotannin (active component of green tea and red wine), isoorientin (A. wardii), isovitexin(A. wardii), neral (M. officinalis), and vitexin (A. wardii) have been identified with medicinal properties and reported useful against different infectious diseases (Table 4, Fig. 6), and hence downloaded from PubChem database of NCBI web server.
Molecular docking analysis
The results of molecular docking were intended for energetically favorable binding poses for each nine natural compounds within the active pocket (LbH domain) of proposed drug target of B. petrii (Fig. 5A) as lowest binding energy and inhibition constant are generally preferred for favorable binding mode while docking small compounds with target protein molecule [42,43]. Again, among them isoorientin (ŌĆō5.66 kcal/mol), curcumin (ŌĆō5.38 kcal/mol), isovitexin (ŌĆō5.22 kcal/mol) found with lower binding energy and inhibition constants i.e., 71.35 ┬Ąm, 114.67 ┬Ąm, and 149.85 ┬Ąm, suggesting as strong inhibitors of lpxD protein. Further, the binding stability was proved through existence of hydrogen bonding pattern between the protein-ligand complexes (Table 5). Presence of hydrophobic amino acid residues within the binding site highly contributes towards the stability during molecular interaction between protein and small compound. Here, the resulted hydrophobic interaction between the docked complexes also supported for a strong binding affinity of natural compounds towards the proposed drug target (lpxD) of B. petrii (Table 6, Fig. 7). Also, the presence of some positively and negatively charged amino acid residues of lpxD within 4 ├ģ distance from the ligands suggested for electrostatic force of interaction between docked complexes (Table 6, Fig. 7). Again, among nine active natural compounds, isoorientin (ŌĆō5.66 kcal/mol) was observed as the most suitable inhibitor with lowest binding energy and inhibition constant (71.35 ┬Ąm), and formed hydrogen bond with amino acid residues like Ala 186, Glu 149, and Gly 227 of lpxD (Table 5, Fig. 7F). Strong hydrophobic interactions were seen due to presence of Val 165, Ala 186, and Leu 226 within 4 ├ģ distance from the ligand (Table 6, Fig. 7F). All of these above observation established isoorientin to be a strong inhibitor against lpxD of B. petrii.
In conclusions, Identification of potential drug target has a vital role in designing a novel drug against any infectious disease. B. petrii strains have been reported as causing infections in humans and various warm blooded animals. In the present investigation, authors attempted to narrow down the complete data set of known 5,031 protein sequences of B. petrii DSM 12804 (accession No. PRJNA28135) in order to identify a potential drug target computationally. In silico study revealed lpxD protein of B. petrii as a potential drug target, is an essential protein of the pathogen and involved in the biosynthesis of the lipid A component of lipopolysaccharide. Again, the observations made through computational study also strongly supported lpxD to be a potential drug target due to the absence of lpxD protein in any human essential pathways as well as without any significant percentage of sequence homology with human and human gut flora micro biota. Again, previous literature reports also established the involvement of lpxD in nosocomial infections caused due to pathogens and hence, could be used as a potential drug target [15,16] against B. petrii. Furthermore, due to adverse effect of synthetic drugs, the uses of active natural compounds have gained an increasing attention towards the treatment or prevention of infectious diseases. Moreover, some of the phytochemicals like ajoene (garlic), allicin (ginger), cinnamaldehyde (Chinese cinnamon), curcumin (turmeric), gallotannin (green tea and red wine), isoorientin (blueberry relative found in Central and South America), isovitexin (blueberry relative found in Central and South America), neral (lemon balm), and vitexin (blueberry relative found in Central and South America) have been reported as having anti-oxidant, anti-inflammatory, and anti-bacterial properties, might be used against COPD, cystic fibrosis, QS biofilm formation, nosocomial infection, and other types of bacterial infection [20,21,22,23,24,25]. Therefore, authors excited to apply molecular docking approach to check the inhibitory effects of above mentioned nine natural compounds (Table 4, Fig. 6) against the proposed drug target (lpxD) of B. petrii. The docking study results also supported for the confirmation of the mechanism of blocking of lipid A biosynthesis in B. petrii which is essential for the survival of the bacterium. Again, isoorientin was noticed with highest inhibitory effect among nine natural compounds supported by lowest binding energy and inhibition constant (Table 5). Binding stability was also confirmed through observation of strong hydrophobic, electrostatic and hydrogen bonding interactions between protein-ligand complexes (Tables 5 and 6, Fig. 7F). Hence, isoorientin could be suggested as most suitable natural compounds against the proposed drug target (lpxD) of B. petrii. The overall hypothesis generated through implementation of in silico approach would be a great aid and value towards designing a novel drug against B. petrii infection and which would be further validated through in vitro experiments.