Tissue Specific Expression Levels of Apoptosis Involved Genes Have Correlations with Codon and Amino Acid Usage
Article information
Abstract
Different mechanisms, including transcriptional and post transcriptional processes, regulate tissue specific expression of genes. In this study, we report differences in gene/protein compositional features between apoptosis involved genes selectively expressed in human tissues. We found some correlations between codon/amino acid usage and tissue specific expression level of genes. The findings can be significant for understanding the translational selection on these features. The selection may play an important role in the differentiation of human tissues and can be considered for future studies in diagnosis of some diseases such as cancer.
Introduction
Regulation of gene expression level is an important step in different biological processes [12]. The expression levels are the overall results of the transcriptional and post-transcriptional regulations [13]. Codon usage bias usually refers to different frequencies of synonymous codons attributed to one specific amino acid [45]. Synonymous codons translate with different speeds; an event which indicates that the translational efficiency may be specifically modified by using different codons [6]. Furthermore, in some prokaryotes and eukaryotes, amino acid content is also known to be related with gene expression level [7]. Two major factors including natural selection and mutational bias are reported to explain such codon and amino acid usage bias [8]. However, this relationship, as proposed by the translation selection model, is less evident in mammals.
There is a clear relation between the expression levels of genes and codon composition in most organisms [5]. Nonetheless, due to tissue-specific gene expression, identification of factors affecting gene expression in multicellular organisms is much more complicated [9]. Thus, some studies have focused on the relation between tissue specific gene expression and compositional features of the genes. Sémon et al. [10] reported that selective pressure influenced the synonymous codon usage of human tissue-specific genes and modify the expression of special proteins. Also, Zhou et al. [9] identified some significant correlations between tissues specific oncogenes and sequence compositional features in human tissues. Then, Hajjari et al. [11] found that the structural elements in tumor suppressor genes and proteins can play important roles in their regulation. However, it is still unclear whether translational selection affects other human genes specifically based on their tissue expression patterns [12].
Apoptosis, the process of programmed cell death, is considered as a crucial component of different processes such as normal cell cycle, development, and function of the immunity system [13]. Different genes controlling the apoptosis process have been identified. Since the expression levels of genes involved in apoptosis are different in human tissues, we aimed to find if the compositional features of these genes are involved in their regulation. Considering the important role of these genes in cellular and molecular function, the results may provide new clues about their tissue specific regulation. Since the dysregulation of these genes is a common event in some diseases especially cancers, the unraveling different mechanisms of their regulation may also reveal novel strategies for therapeutic intervention.
In this study, we tried to elucidate the potential correlation between some compositional features of genes/proteins structures and their expression level in some human tissues. The results may reveal the support for the relation between codon usage, amino acid usage and tissue specific gene expression.
Methods
Sequence retrieval, alignments, and expression data acquisition
The list of apoptosis involved genes coding proteins with different molecular functions and localizations in the cell were drawn from the Deathbase (http://deathbase.org/) (Supplementary Table 1). In order to minimize the statistical errors, multiple alignments were performed for the sequences by ClustalW program (http://www.ebi.ac.uk), and finally, 65 genes were selected for our study. Normalized expressions of genes were directly drawn from SOURCE, 2014 (Stanford Microarray) database (Source.stanford.edu) (Supplementary Table 1). The normalized gene expression in this database presents the relative expression level of a gene (defined as a UniGene Cluster) in different tissues and is “normalized” for the number of clones from each tissue that are included in UniGene (http://source-search.princeton.edu/help/SOURCE/normalization.html). This database links to the microarray experiments that included the queried genes. Thirty four different tissues were selected and in each tissue, the average expression level, the number of expressed genes, and the highest expressed gene were recognized.
Sequence compositional features
The codon usage table of Homo sapiens was obtained (http://www.kazusa.or.jp/codon/), and used to calculate Codon Adaptation Index (CAI) (http://genomes.urv.es/CAIcal/) for each coding domain sequence (CDS). The tRNA Adaptation Index (tAI) was also measured for each CDS using Visual Gene Developer 1.4 Build 750. The frequencies per thousand of all of 61 codons were calculated for all 65 CDSs by Countcodon program (http://www.kazusa.or.jp/codon/countcodon.html). Also, The Relative Synonymous Codon Usage (RSCU) of all amino acids was measured (http://www.bioinformatics.org/sms2/codon_usage) for all desired proteins.
Amino acid sequence characteristics
The protein sequences of all 65 genes were obtained from NCBI Resource Portal. Then, the percentage of each amino acid was obtained for all of the proteins using Protein Information Resource (http://pir.georgetown.edu).
Statistical analyses
For each tissue, the correlation between gene expression level of desired genes and compositional features of CDS/protein was analyzed by Graphpad. p-values less than 0.01 were considered as significant. However, in some tissues, because of lacking the p-value less than 0.01, we considered the characteristics with p-values less than 0.05 as the most significant features. Finally, in order to determine truly significant features, false discovery rate (FDR) was analyzed through the FDR online calculator (http://www.sdmproject.com/utilities/?show=FDR) which its method coincides with the R code of the version proposed by Benjamini and Hochberg.
Results
Correlation between gene compositional features and expression level
The correlation analyses showed that the gene compositional features are associated with the expression levels of desired genes. The results indicated that the expression levels have significant correlation with the frequencies of some codons such as AAG (in 20 tissues), AUC (in 17 tissues), and GAC (in 12 tissues) (Supplementary Table 2). Among these codons, AAG and AUC showed the most significant correlation coefficients. In order to identify the tissues in which both AAG and AUC have significant correlations with the gene expression level, a bar plot was drawn by R software for these codons (Fig. 1). In Supplementary Table 2, we can see that the most frequent and attributed tRNAs are attributed to the AAG-Lys, AUC-Ile and GAC-Asn codons.
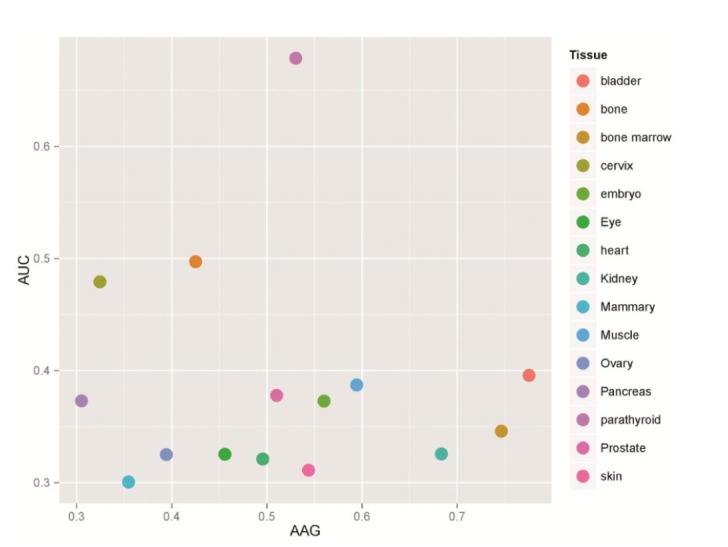
The tissues in which both AAG and AUC have significant correlations with the gene expression level. The bar plot is drawn by the R statistical software. p < 0.05 is considered statistically significant.
Furthermore, our data showed that the expression levels of apoptosis genes have significant correlations with the relative synonymous codon usage features such as CCC (Prolin) and TCC (Serine) (Supplementary Table 3). Since the aforementioned codons have the most significant correlation coefficients, a bar plot was drawn by R software for these features in order to identify the tissues in which both codons have significant correlation coefficients with gene expression (Fig. 2).
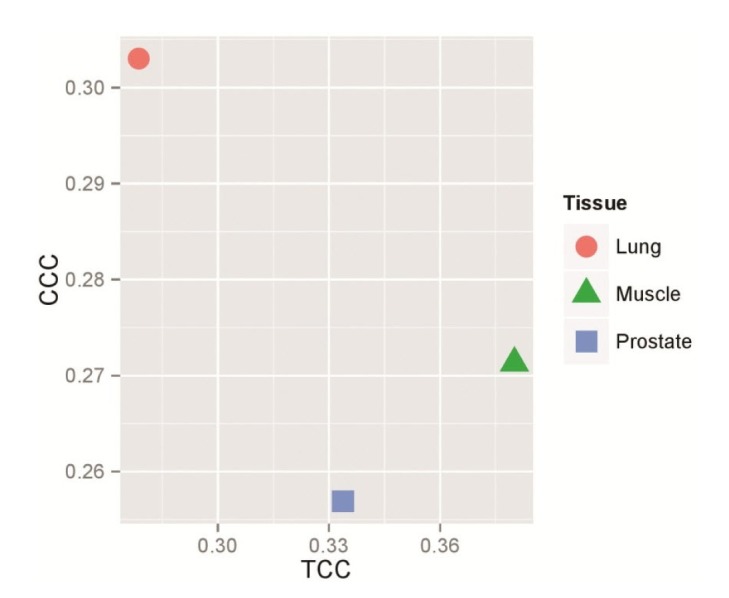
The tissues in which the codons CCC (proline) and TCC (serine) have the most significant correlation coefficients with gene expression levels. The bar plot is drawn by R statistical software. p < 0.05 is considered statistically significant.
To find the level of codon bias, CAI for each gene was measured. We found some correlations between these parameters and the expression level of desired genes in 8 tissues (p < 0.05). Furthermore, we calculated the tAI, which is a measure of the tRNA usage by coding sequences. Significant correlations between tAI and the expression level of genes were reported in 20 tissues (Supplementary Table 4). Also, the results indicated that nucleotide compositional features (including CG percent in codon bases) have significant correlations with gene expression levels in some tissues (Supplementary Table 4).
Correlation between protein compositional features and the expression levels
Our data indicated that different amino acids including Arg, Asp, Lys, Glu, Gln, Ser, and Ile have significant correlations with the expression level of genes in 15, 14, 12, 11, 11, 9, and 8 tissues respectively (Supplementary Table 5). Among these notable amino acids, Asp and Lys have the most significant positive correlation coefficients. So, a bar plot was drawn by R software for these amino acids in order to identify the tissues in which both Asp and Lys have significant correlation coefficients with gene expression level (Fig. 3).

The tissues in which both Asp and Lys have the most significant positive correlation coefficients with gene expression levels. The bar plot is drawn by R statistical software. p < 0.05 is considered statistically significant.
Most significant and truly significant features in different tissues
In order to find out which feature has the most significant correlation with the gene expression level in different tissues, the features with the least p-value were recognized (Table 1, Fig. 4). Also, since multiple correlations were done in the current study, the FDR analysis was done to decrease the statistical errors (Table 2). Through this analysis, we calculated the corrected p-values in order to find truly significant feature. By FDR analysis, we found some truly significant features such as AAG (coding lysine) and GAC (coding aspartate) codons (drawn from codon frequencies), lysine and aspartate (drawn from amino acid frequencies), and CTCL (from synonymous codon usage features) which are common among some tissues (Table 2).
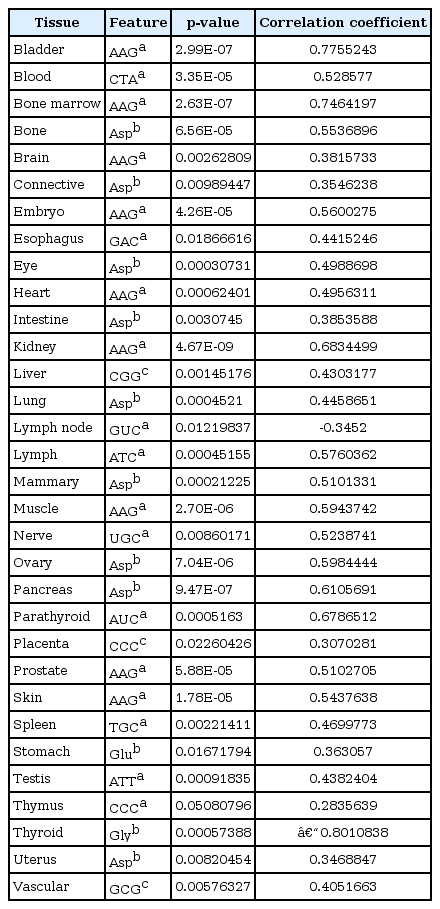
Features with the most significant correlation with gene expression levels in different tissues

The features which have the most significant correlations with gene expression levels in different tissues. The scatter plot is drawn by the R statistical software.
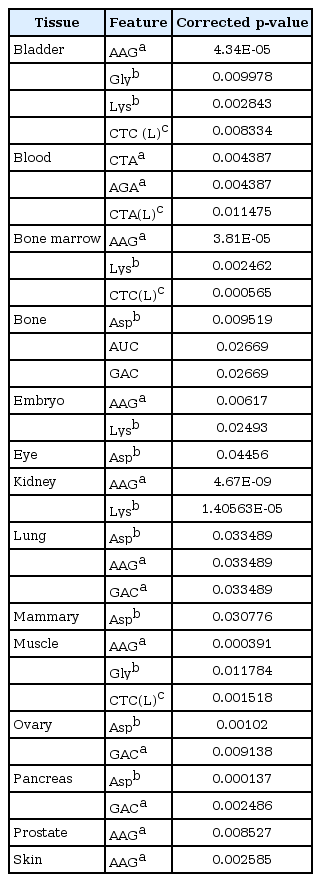
The FDR corrected features which have the correlation with genes expression levels
Discussion
The relation between the expression level and the gene/protein compositional features has been reported in different organisms [5]. However, little is known about the factors affecting the tissue specific gene expression level in multicellular organisms [9]. In the last years, the effects of codon bias and other compositional features on the tissue-specific translational control of genes have been reported [111]. In the current study, in order to elucidate this level of regulation on the expression of apoptosis involved genes, we investigated the relation between the gene expression and structural features. The results can help us understand the molecular mechanism and regulation of this type of genes. Our data suggest that synonymous codon usage in human genes may not be the result of isochore organization of the genome or neutral evolutionary processes as well. We found some common features correlated with gene expression in different tissues, a finding that may indicate common mechanisms in gene regulation. The current results suggest several hypotheses about the mechanisms of gene regulation and tissue differentiation in human.
There are some reports indicating that codon usage in mammals has notable effects on translation rate [14], especially during cell differentiation [15]. The systematic tissue-specific codon usage may indicate that human tissues can differ in the relative tRNA amount [16]. This may influence on the expression of the desired proteins. The significant correlation between gene expression level and some codon frequencies especially AAG codon (truly significant in nine tissues) may indicate the importance of the abundance of attributed tRNAs pairing with this codon. The correlation between gene expression and frequency of AAG codon was interestingly approved in a previous study by Hajjari et al. [11] on Tumor suppressor genes.
Our data can support the Plotkin et al.'s study on synonymous codon usage features [3]. Our data on the significant correlations between tAI and expression levels of genes may support this hypothesis. The relation between synonymous codon usage bias and gene expression levels might be the result of the weak selection on synonymous sites which generate translationally optimal codons [1718]. Among our data, the correlation between the synonymous codon usage of leucin and gene expression level is of noted. In a similar study on tumor suppressor genes, this type of correlation was also observed [11]. So, these data altogether may indicate the importance of synonymous codon usage of Leucine in the structure of genes. Altogether, some correlations between relative synonymous codon usage, codon frequency, tAI, CAI, and gene expression show that there may be a translational selection on apoptosis involved genes. Since some of these features are common between different tissues, we assume that common mechanisms may be involved in some specific tissues in this level of regulation.
Some previous studies declare that the amino acid composition of proteins varies with increasing levels of gene expression in different tissues [192021]. This result might suggest translational selection at the amino acid level. Based on this, the amino acids that are used more frequently in highly expressed genes may correspond to the most abundant tRNAs [1922]. Furthermore, some studies also demonstrate that amino acid composition is affected by selection to decrease the metabolic cost of protein production [23]. In this study, we showed tissue specific correlations between amino acid usage and expression levels of apoptosis genes. Altogether, it may be inferred that the content of amino acids in tissues are very important for the regulation of the expression level of apoptosis involved genes. According to our results, some amino acids have tissue specific correlations with the expression of apoptosis genes. Our results showed that different amino acids including Arg, Asp, Lys, Glu, Gln, Ser, and Ile have significant correlations with the expression level of genes in 15, 14, 12, 11, 11, 9, and 8 tissues respectively (Supplementary Table 5). If we consider all of the codons attributed to one specific amino acid in one group for correlation analysis, we also get the same correlation coefficient and p-value as the amino acid features. So, all of the codons of arginine including CGT, CGA, CGC, CGG, AGA, and AGG (as one group) has the significant correlation with gene expression level. Some previous studies have demonstrated the role of these amino acids in apoptosis progression [2425]. So, it seems that the concentration of these amino acids in specific cell types can play an important role in cell survival and apoptosis.
Altogether, our results demonstrate the potential translational selection on sequence features of human apoptosis genes. Our data support the previous studies performed by Hajjari et al. [11] and Zhou et al. [9] who showed that the expression of cancer involved genes are correlated with compositional features in different human tissues. The current findings have implications for the optimal design of gene therapies targeted in specific tissues and will promote a better biological understanding of translational selection of tissue specific gene expression in human.
Acknowledgments
We acknowledge Shahid Chamran University of Ahvaz for supporting this study.
References
Supplementary materials
Supplementary data including five tables can be found with this article online at http://www.genominfo.org/src/sm/gni-14-234-s001.pdf.
Supplementray Table 1
List of the genes analyzed in the study
Supplementary Table 2
The coefficients of codon frequencies which have the significant correlations with genes expression levels in different tissues
Supplementary Table 3
The coefficients of relative synonymous codon usage features which have the significant correlations with genes expression levels in different tissues
Supplementary Table 4
Correlation coefficients between tAI/CAI and genes expression levels in different human tissues
Supplementary Table 5
The coefficients of amino acid frequencies which have the significant correlations with genes expression levels in different tissues