Investigation of Splicing Quantitative Trait Loci in Arabidopsis thaliana
Article information
Abstract
The alteration of alternative splicing patterns has an effect on the quantification of functional proteins, leading to phenotype variation. The splicing quantitative trait locus (sQTL) is one of the main genetic elements affecting splicing patterns. Here, we report the results of genome-wide sQTLs across 141 strains of Arabidopsis thaliana with publicly available next generation sequencing datasets. As a result, we found 1,694 candidate sQTLs in Arabidopsis thaliana at a false discovery rate of 0.01. Furthermore, among the candidate sQTLs, we found 25 sQTLs that overlapped with the list of previously examined trait-associated single nucleotide polymorphisms (SNPs). In summary, this sQTL analysis provides new insight into genetic elements affecting alternative splicing patterns in Arabidopsis thaliana and the mechanism of previously reported trait-associated SNPs.
Introduction
Arabidopsis thaliana is a metaphyte and angiosperm growing in Europe, Asia, and Africa. This is a small plant that is one of the widely popular model organisms, because this plant has a small genome and can be cultivated easily. Thus, much genetic research has been carried out with Arabidopsis thaliana [12]. Recently, in Arabidopsis thaliana, genetic components associated with traits or diseases have been reported through genome-wide association studies (GWAS) studies [3]. The GWAS is a powerful approach to discover genetic elements associated with various traits. The GWAS was based on 107 phenotypes. This can be grouped into 4 categories: 23 flowering, 23 defense, 18 ionomics, and 43 development traits. Although GWASs have been successful, the molecular mechanisms of genetic elements discovered through the GWAS have been elusive [4]. The molecular mechanisms of previously reported trait-associated single nucleotide polymorphisms (SNPs) in Arabidopsis thaliana have been also elusive. Expression quantitative trait loci (eQTLs) influencing gene expression levels and splicing quantitative trait loci (sQTLs) changing alternative splicing patterns are useful genetic elements to explain the trait-associated SNPs. In Arabidopsis thaliana, there have been many studies to identify eQTLs, and these accumulated eQTLs have been greatly helpful for explaining trait-associated loci [567]. However, there have been few studies of the genome-wide investigation of sQTLs in Arabidopsis thaliana. Since sQTLs can change alternative splicing patterns and consequently affect various traits, those sQTLs can constitute crucial elements for the interpretation of trait-associated SNPs [8]. Here, we discover candidate genome-wide sQTLs in Arabidopsis thaliana using the IVAS package, which is a user-friendly R/Bioconductor package, to discover sQTLs with genotypes and fragments per kilobase million (FPKM) of a transcript dataset [9]. We analyzed 141 previously published matched genotype data and an RNA-seq dataset downloaded in the 1001 Genomes Data Center [10] and the Gene Expression Omnibus (GEO) database [11], respectively. To obtain the FPKM of transcripts, we processed and aligned raw RNA-seq data. As a result of the discovery using IVAS, we identified 1,694 SNPs. Furthermore, among those SNPs, we found 96 candidate sQTLs that overlapped with the list of previously published trait-associated loci [3], and of 96 sQTLs, 25 sQTLs that led to a large difference in expression ratio of alternatively targeted exons across genotypes of each sQTL were finally selected. In summary, we reported 1,694 genome-wide sQTLs and 25 candidate trait-associated sQTLs in Arabidopsis thaliana. This list of those sQTLs may be a useful dataset for sQTL utilities in Arabidopsis thaliana, as well as its neighboring species.
Methods
The overall pipeline
An overview of our research is given in Fig. 1. We downloaded genotype and RNA-seq datasets measured in Arabidopsis thaliana from the 1001 Genomes Data Center [10] and the GEO database (GSE43858) [11]. The genotype and RNA-seq datasets include 171 and 160 strains, respectively. Among them, we selected a matched dataset of 141 strains. The raw RNA-seq data were filtered by quality control and mapped to a reference sequence using SHRiMP2 software, which is an alignment tool for high accuracy and sensitivity at a very reasonable speed [12]. An average mapping rate was about 80%. After mapping to the reference sequence, we filtered out mapped reads with low mapping quality using Samtools [13]. In order to obtain FPKM values, we estimated quantifications from mapped reads using Cufflinks [14] with the reference GTF file downloaded from the Ensembl Genomes website. With the preprocessed genotype and RNA-seq datasets, we carried out IVAS for the discovery of genome-wide sQTLs. Afterwards, we found candidate sQTLs that overlapped with trait-associated loci in the list of the 107 phenotypes database [3].
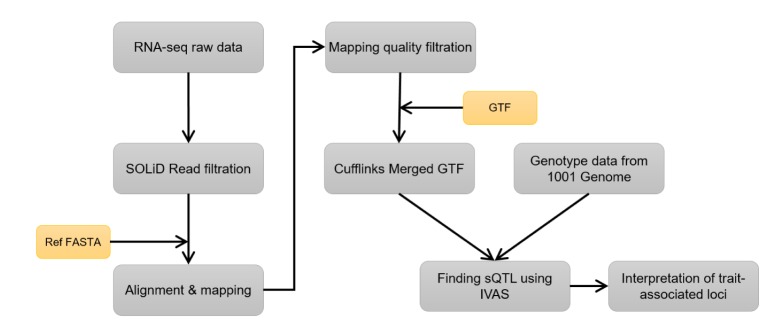
The overview of our analysis. The RNA-seq raw reads were filtered by a quality score, and then, the filtered reads were mapped to a reference genome using SHRiMP2. After mapping, filtration with mapping quality was carried out using Samtools. The filtered mapped reads and genotype data were analyzed by IVAS in order to obtain the candidate splicing quantitative trait loci.
Association test for identification of genome-wide sQTLs in Arabidopsis thaliana
In order to discover genome-wide sQTLs in Arabidopsis thaliana, we carried out an association test using the IVAS package [9]. For each exon with at least two isoforms, IVAS tested associations between the alternatively targeted exon and the genotype of SNPs located in the exon and flanking introns. We filtered the result based on a cutoff at false discovery rate (FDR) = 0.01 and median values of exon ratio across each genotype > 0.1.
Interpretation of trait-associated SNPs with candidate sQTLs
We downloaded the list of loci associated with the 107 phenotypes of 4 categories [3] (p < 0.05). We identified and tabulated the trait-associated loci overlapping with the candidate sQTLs obtained by IVAS. In order to yield a more biologically meaningful result, we kept those trait-associated sQTLs achieving a high differential expression ratio of an alternatively targeted exon across genotypes of each sQTL (the third quantile of exon ratios of one genotype > the first quantile of exon ratios of the other genotype).
Results
Processing for the genotype and RNA-seq datasets
We downloaded a genotype dataset in 1001 Genomes Data Center [10] and an RNA-seq dataset in the GEO database (GSE43858) [11] in order to identify genome-wide sQTLs in Arabidopsis thaliana. In order to increase the accuracy of the mapping of RNA-seq reads, we manually filtered out low-quality reads and aligned them to the Arabidopsis thaliana reference genome. The aligned reads were estimated by Cufflinks [14]. As a result, we obtained and analyzed the quantifications of 41,621 isoforms and 5,833,535 SNP markers of the 141 matched strains.
Discovery of sQTLs in Arabidopsis thaliana
Using the IVAS package [9], we analyzed the preprocessed dataset. For each alternatively spliced exon, we carried out an association test between a given exon expression and genotype of an SNP located in the given exon and flanking introns. As a result, we found 1,694 candidate sQTLs by a cutoff at an FDR = 0.01. Fig. 2A displays an exemplary boxplot of an sQTL, located in chr3:20,536,243, which shows the association between its genotype and an expression ratio of the AT3G55400 ninth exon.
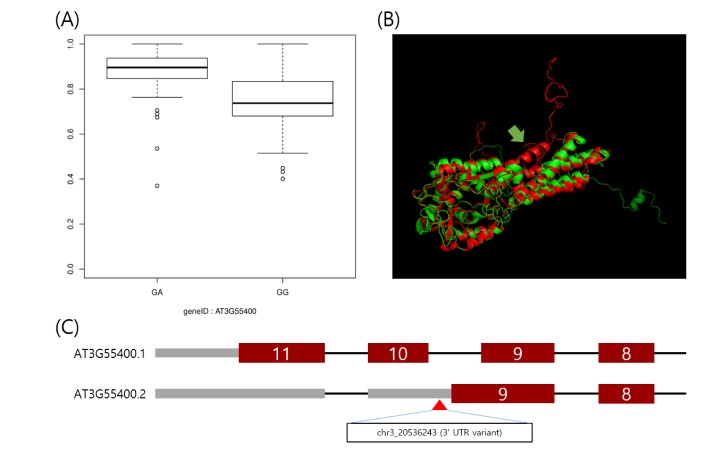
Example of the utility of splicing quantitative trait loci for interpretation of trait-associated loci. (A) Boxplot of expression ratio of the exon with alternative 3-prime splicing site between phenotypes of the polymorphism, located in chr3:20536243. The expression ratio is higher in the heterozygote. (B) This is the predicted protein structure of the AT3G55400 gene using the RaptorX web-based tool [16]. The alternative 3-prime splicing site leads to the loss of alpha helices (yellow arrow). (C) The single nucleotide polymorphism, a 3′-untranslated region variant, likely leads to an alternative 3′ splicing site.
The sQTLs and trait-associated SNPs
Here, we interpreted the molecular mechanism of previously published trait-associated SNPs into sQTLs. The published trait-associated SNPs were downloaded from the 107 phenotypes database [3]. This database includes 107 phenotypes of 4 categories. We looked for the sQTLs that overlapped with SNPs in the database resource and selected SNPs having a significant association with phenotypes. As a result, we found 96 candidate sQTLs that were able to explain the mechanism of trait-associated SNPs. Among the 96 candidate sQTLs, we finally selected 25 sQTLs affecting high differential expression of an alternatively spliced target exon across genotypes. We show an example of a trait-associated sQTL in Fig. 2. The SNP, which resides in chr3:20,536,243, affects the ninth exon of AT3G55400, OVA1. That is, the expression ratio of the exon with the alternative 3′ splice site is higher in the heterozygote (AG) than in the homozygote (GG). OVA1 encodes a methionine-tRNA ligase and has an association with ovule development, and its polymorphism was reported to be associated with the pedicel [3]. We report the full list of trait-associated sQTLs in Table 1.
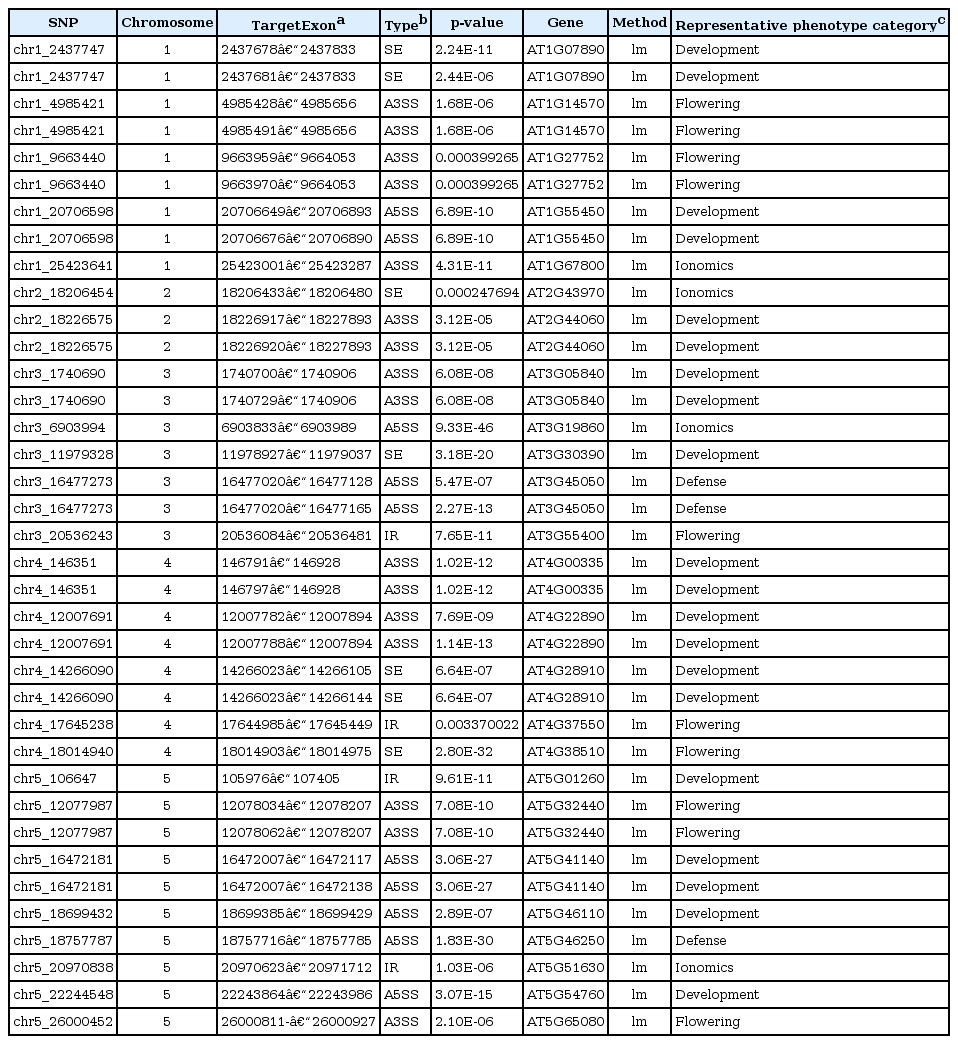
The candidate trait-associated sQTLs
Discussion
Here, we report 1,694 genome-wide sQTLs in Arabidopsis thaliana with a high-throughput RNA-seq dataset. RNA-seq using next generation sequencing provides an accurate method for annotation and quantification of exons than the exon array method, and this is greatly helpful for the investigation of sQTLs [15]. Arabidopsis thaliana is a model organism to study the genetic elements of plants [1]. Thus, an sQTL survey in Arabidopsis thaliana as a model organism has importance for understanding the regulation of alternative splicing of plants. However, there are only a few studies on sQTLs in plant. Thus, our result may be a great resource to explain the genetic regulation of alternative splicing patterns. Furthermore, the sQTLs can be useful for explaining known trait-associated SNPs. A recent study showed that genetic markers with various traits have been discovered in Arabidopsis [3]. However, their molecular mechanisms have remained elusive. sQTLs are able to alter alternative splicing patterns, and consequently, they can lead to phenotypic variations [5678]. Thus, our result can be useful in interpreting trait-associated SNPs as sQTLs. For example, the OVA1 gene encodes a functional protein for methionine-tRNA ligase, having several alpha-helices. The polymorphism, in chr3:20,536,243, was reported to have an association with the flower pedicel [3]. However, this molecular mechanism has been not elucidated. Through our result, the SNP was shown to have an association with an alternative 3′ splicing site of the ninth exon of the OVA1 gene. We predicted the three-dimensional structure of proteins translated by transcription, including normal exons and the alternative 3′ splicing site of the ninth exon, respectively, using the RaptorX Structure Prediction tool, a web-based protein structure prediction tool [16]. As a result, translation of the transcript with the alternative 3′ splicing site showed a loss of 2 alpha helices (Fig. 2B). It is tempting to speculate that the SNP, in chr3:20,536,243, which is known as a trait-associated locus, is able to lead to translation of abnormal protein and consequently affect the flower pedicel. Further biochemical studies of this alternative spliced transcription may be able to validate our prediction. Our result can explain only a small portion of trait-associated SNPs. However, these sQTLs can be a powerful resource for the interpretation of more trait-associated loci that will be found in the coming years. Furthermore, since there have been few investigations of genome-wide sQTLs in Arabidopsis thaliana, our result can be a reference resource of sQTLs for its neighboring species.
Acknowledgments
The financial support of this work was made available by the National Research Foundation of Korea (NRF-2012M-3A9D1054705), funded by the Ministry of Education, Science, and Technology, and by the Rural Development Administration of Korea (PJ01167402).