Introduction
The meta-analysis is a tool for pooling information from multiple independent studies [1,2,3,4]. In the field of genetics, the meta-analysis has become a popular way of aggregating information from multiple genome-wide association studies (GWASs) in order to increase statistical power while controlling for the rate of false positive findings [5,6,7,8,9,10,11,12,13]. The meta-analysis has also become a useful tool for many applications of bioinformatics, such as neuroimage processing [14] and expression quantitative trait loci analysis [15].
There exist several approaches for combining information from multiple studies. Statistical methods can differ depending on the scenario: when (1) test statistics are unknown but only p-values are available, (2) test statistics are known but data are not available, or (3) actual data are available. In this paper, we focus on scenario (2), which is a common situation in genetic studies. We note that for scenario (1), Fisher's method for combining p-values is commonly used [16]. In scenario (3), we can combine actual data, which is rarely doable in retrospective studies or in genetic studies where transferring genotype data is difficult due to privacy issues. For scenario (2), which we focus on, the fixed effects model meta-analysis is the most common approach for synthesizing test statistics from multiple studies [1,17].
To perform a fixed effects model meta-analysis, there are two popular methods: the inverse variance-weighted average and the weighted sum of z-scores (SZ) [2,17,18]. The inverse variance-weighted average method (IVW) summarizes effect sizes from multiple independent studies by calculating the weighted mean of the effect sizes using the inverse variance of the individual studies as weights. The weighted SZ method constructs a new z-score by calculating a weighted sum of individual z-scores. It has been known that the sample size of individual studies is a preferable weight for the method [10,19,20]. Although several empirical evidence has shown that the two methods perform similarly [2,17,21], the characteristics of each method and the analytical connection between the two methods have not been thoroughly investigated.
In this paper, we first investigate the optimal characteristics of the two methods. We show that the two methods are optimized for different optimality criteria: IVW maximizes the likelihood function, which is equivalent to minimizing the estimator variance, and SZ maximizes the non-centrality parameter of the statistic, which is equivalent to maximizing the statistical power. This characterization gives us insight into the optimal weight for SZ; using only the sample size information as weights can often be suboptimal in terms of statistical power compared with using all information as weights, such as minor allele frequencies. Although the two methods are optimized for different goals, we analytically demonstrate that the two methods become equivalent under certain assumptions that hold over a wide range of applications. We examine this connection between the two methods both analytically and empirically. Finally, using real data analysis utilizing the Wellcome Trust Case Control Consortium data, we demonstrate that the two methods can give distinct results in certain study designs.
Methods
Inverse variance-weighted average method
We first describe the two methods for the fixed effects model meta-analysis: the IVW and weighted SZ. The fixed effects model assumes that all studies in a meta-analysis share a single true effect size [2,18,22,23]. The underlying mathematical model of the observed effect Xi can be shown as: where ┬Ą is the true effect size and ei (the deviation of Xi from ┬Ą) is the error in the observation and i = 1,2,ŌĆ”,C. In order to integrate multiple observed effect sizes X1,ŌĆ”,XC from multiple studies, the weighted mean approach has been suggested [22],
A choice of weight Wi is not immediately evident, but several attempts were made to identify the optimal weight of the methods based on empirical evidence [17,20,24]. Ideally, one needs to put more weight on the studies with more precision against studies with lower precision [3,25,26]. When the sample size of each study is sufficiently large, we can assume that Xi follows a normal distribution approximately, based on the central limit theorem. This applies to situations where the data themselves are not normal (e.g., binary), in which situation the test statistic still follows a normal distribution, as long as the sample size is large. In GWASs, this assumption holds easily, because the sample size is typically as large as thousands of samples. Note that all derivations in this paper are based on this normality assumption. Let SE(Xi) be the estimated standard error of Xi, and Vi = SE(Xi)2. It is common practice to consider the estimated variance Vi as the true variance. The inverse variance-weighted average effect size estimator is the weighted mean of Xi with the weights [22]:
Given these weights, the standard error of the average effect size X ¯ 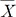 becomes
becomes S E X ┬» = Ōłæ W i ŌłÆ 1 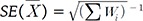 . The statistical significance can be tested by constructing a z-score statistic of IVW as follows, which asymptotically follows N(0,1) under the null hypothesis of no effects.
. The statistical significance can be tested by constructing a z-score statistic of IVW as follows, which asymptotically follows N(0,1) under the null hypothesis of no effects.
becomes . The statistical significance can be tested by constructing a z-score statistic of IVW as follows, which asymptotically follows N(0,1) under the null hypothesis of no effects.Results
Below, we show the characteristics of the two methods and the connections between the two methods. We first show that each method is optimized to meet a unique optimality criterion. Then, we show that the two methods are connected, by using both analytical derivations and empirical simulations. Finally, we demonstrate a situation in which the two methods can give different results using real data.
Optimality of IVW
IVW maximizes likelihood function
We will define that a method is optimal if the method achieves a specific goal more effectively than any other method. We show that IVW is optimal in two different aspects: (1) the summary estimator gives the greatest likelihood than any other estimator and (2) the summary estimator's variance is smaller than the variance of any other estimator. First, we show that IVW is optimal in the sense that the IVW estimator maximizes the likelihood function. Suppose that we have a series of n studies with observed effect sizes Xi, i = 1, 2, ŌĆ” n. Under the fixed effects assumption, there exists a true effect size ┬Ą, and each observation Xi comes from a normal distribution with mean ┬Ą and a standard deviation Žāi. The probability density function of each observation is given by
Because ŌłÆlnL(┬Ą,Žāi2|X1,ŌĆ”,Xn) will be minimized at d ( ŌłÆ l n L ┬Ą , ŌĆć Žā i 2 X 1 , ŌĆć ŌĆ” , ŌĆć X n ) d ┬Ą = 0 ŌĆć 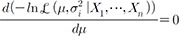 , we can obtain the maximum likelihood (ML) estimator: which is equivalent to the IVW statistic
, we can obtain the maximum likelihood (ML) estimator: which is equivalent to the IVW statistic X ¯
, we can obtain the maximum likelihood (ML) estimator: which is equivalent to the IVW statistic in equation (2). Therefore, the inverse variance-weighted average method is optimal in the sense that it maximizes the likelihood of observations with the optimized weight of Wi = (Žāi2)ŌłÆ1 [21,22].IVW achieves minimum variance
IVW is optimal in the sense that the IVW estimator achieves minimum variance. In short, IVW achieves minimum variance by the properties of maximum likelihood estimator (MLE). MLE has the following property, as shown by Greene [27]: if the sampling is from an exponential family of distributions and the minimum variance unbiased estimator (MVUE) exists, that estimator becomes the ML estimator [27]. Thus, by this property and under these conditions, we can conclude that IVW achieves minimum variance, because IVW is the MLE and MVUE [27].
Optimality of SZ
SZ maximizes the non-centrality parameter
SZ combines z-scores from multiple studies to construct a new z-score. Therefore, in SZ, we are not interested in the estimator of Xi. Rather, we are interested in the statistical significance of the combined information. Thus, the goal of SZ is to maximize how much the z-score will be shifted from 0 on average, which is often called the non-centrality parameter. By maximizing the non-centrality parameter, we can maximize the statistical power of the test. Among all possible weights that can construct a weighted SZ, we want to find the weights that will maximize the non-centrality parameter.
The optimal weights of the weighted SZ can be found by the Cauchy-Schwarz inequality. We will make an assumption that
That is, we assume that our z-score is defined as the effect size estimate divided by the standard error, which is the common definition of a z-score. In some applications, there can be different ways to define a z-score statistic, and for those definitions, the connection between z-score and effect size may not be apparent. However, in practice, this assumption holds approximately over a wide range of applications. Below, we will show that in the situation of a 2 ├Ś 2 table, even if we obtain a z-score in a different way, it approximates a z-score that is obtained by using effect size and its standard error.
Under the fixed effect model assumption that assumes E[Xi] = ┬Ą, the z-score, Zi, follows a normal distribution ZSZ~N(╬╗,1), where ╬╗ is a non-centrality parameter with ╬╗ = Ōłæ i = 1 n w S Z , ŌĆć i ( ┬Ą / V i ) / Ōłæ i = 1 n w S Z , ŌĆć i 2 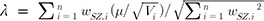 . Now, we want to obtain the weight wi that maximizes lambda. The optimal weight can be obtained by using the Cauchy-Schwarz inequality, as follows [12,21]:
. Now, we want to obtain the weight wi that maximizes lambda. The optimal weight can be obtained by using the Cauchy-Schwarz inequality, as follows [12,21]:
. Now, we want to obtain the weight wi that maximizes lambda. The optimal weight can be obtained by using the Cauchy-Schwarz inequality, as follows [12,21]:
The equality is achieved when where the terms Ōłæ i = 1 n w S Z , ŌĆć i 2  and k are constants. Thus, the optimal weight is
and k are constants. Thus, the optimal weight is w S Z , ŌĆć i = 1 / V i 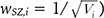 . Then, the resulting weighted SZ can be constructed as
. Then, the resulting weighted SZ can be constructed as
and k are constants. Thus, the optimal weight is . Then, the resulting weighted SZ can be constructed as
This result provides us with the intuition that SZ is optimal only when we weight z-scores by the inverse of the standard errors of effect sizes. That is why previous studies have weighted z-scores by N i 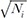 , because in many applications, the variance Vi is inversely proportional to the sample size Ni. However, we would like to note that in some applications, the variance can be a function of not only Ni but also other properties of the data. For example, in genetic association studies, when we test an association of a single-nucleotide polymorphism (SNP) to a phenotype, the variance is typically inversely proportional to Nipi(1ŌłÆpi), where pi denotes the allele frequency of the risk allele. This suggests that if the datasets that we want to combine have different allele frequencies, weighting the z-scores only by Ni can be suboptimal. Below, we will show by simulations that we can have some power loss by using just Ni as the weight, instead of accounting for frequency differences. However, the approximation of this weight
, because in many applications, the variance Vi is inversely proportional to the sample size Ni. However, we would like to note that in some applications, the variance can be a function of not only Ni but also other properties of the data. For example, in genetic association studies, when we test an association of a single-nucleotide polymorphism (SNP) to a phenotype, the variance is typically inversely proportional to Nipi(1ŌłÆpi), where pi denotes the allele frequency of the risk allele. This suggests that if the datasets that we want to combine have different allele frequencies, weighting the z-scores only by Ni can be suboptimal. Below, we will show by simulations that we can have some power loss by using just Ni as the weight, instead of accounting for frequency differences. However, the approximation of this weight N i
, because in many applications, the variance Vi is inversely proportional to the sample size Ni. However, we would like to note that in some applications, the variance can be a function of not only Ni but also other properties of the data. For example, in genetic association studies, when we test an association of a single-nucleotide polymorphism (SNP) to a phenotype, the variance is typically inversely proportional to Nipi(1ŌłÆpi), where pi denotes the allele frequency of the risk allele. This suggests that if the datasets that we want to combine have different allele frequencies, weighting the z-scores only by Ni can be suboptimal. Below, we will show by simulations that we can have some power loss by using just Ni as the weight, instead of accounting for frequency differences. However, the approximation of this weight will be optimal when no heterogeneity can be found between the minor allele frequencies [2].Equality of IVW and SZ under certain assumptions
Analytical derivation
Here, we show that the two methods IVW and SZ are equivalent under certain assumptions. We have shown that IVW is optimal in the sense that the estimator is MLE and achieves minimum variance, and SZ is optimal in the sense that it maximizes the non-centrality parameter. Although both methods can be considered optimal, their goals and how they are optimized are completely different; IVW aims to obtain the best summary estimate (thus MLE and minimum variance), and SZ aims to maximize the statistical power, without considering the summary estimator. Despite the fact that the two methods are optimized differently with different goals, we show that the resulting statistics are equivalent, in the sense that their z-scores (and therefore their p-values) are equivalent. Again, we assume the definition of z-score Z i = X i V i 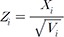 , which is assumed in Eq. (18). We assume that SZ uses SE(Xi)ŌłÆ1 as weights for z-scores, rather than only using sample sizes.
, which is assumed in Eq. (18). We assume that SZ uses SE(Xi)ŌłÆ1 as weights for z-scores, rather than only using sample sizes.
, which is assumed in Eq. (18). We assume that SZ uses SE(Xi)ŌłÆ1 as weights for z-scores, rather than only using sample sizes.Empirical simulation
To empirically investigate the equality of IVW and SZ, we compared the power of the two methods. We assumed the following null and alternative hypotheses:
That is, we tested if the mean effect is non-zero.
To generate simulation sets of meta-analysis studies, we used the common simulation framework for simulating genetic association studies. We assumed that there is a single SNP whose minor allele confers risk of a disease, which is a dichotomous trait. We assumed a number of different relative risks, ╬│ = P d i s e a s e S N P m i n o r P d i s e a s e S N P m i n o r 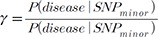 , ranging from 1.01 to 1.2. We assumed a meta-analysis of 5 genetic association studies and assumed a minor allele frequency (MAF) of 0.3. In an additional simulation setting, we assumed varying MAFs of (0.1,0.2,0.3,0.4,0.5) for the 5 studies. We assumed a very small disease prevalence (F Ōēł 0). Given these assumptions and parameters, we can calculate the expected MAF in cases and in controls. Specifically, given MAF p and relative risk ╬│, the case MAF becomes ╬│p/((╬│ŌłÆ1)p + 1), where the control MAF becomes approximately p, given F Ōēł 0. Given the expected MAF in cases and controls, we could randomly sample genotype data, assuming 500 cases and 500 controls for each of the five studies. To assess the statistical significance of the sample data, we used log odds ratio as a statistic, which follows an asymptotic normal distribution. We repeated the procedure to generate 100,000 simulated meta-analysis sets. Given the significance level ╬▒ = 0.05, the power was the proportion of sample sets whose meta-analysis p-value was Ōēż╬▒.
, ranging from 1.01 to 1.2. We assumed a meta-analysis of 5 genetic association studies and assumed a minor allele frequency (MAF) of 0.3. In an additional simulation setting, we assumed varying MAFs of (0.1,0.2,0.3,0.4,0.5) for the 5 studies. We assumed a very small disease prevalence (F Ōēł 0). Given these assumptions and parameters, we can calculate the expected MAF in cases and in controls. Specifically, given MAF p and relative risk ╬│, the case MAF becomes ╬│p/((╬│ŌłÆ1)p + 1), where the control MAF becomes approximately p, given F Ōēł 0. Given the expected MAF in cases and controls, we could randomly sample genotype data, assuming 500 cases and 500 controls for each of the five studies. To assess the statistical significance of the sample data, we used log odds ratio as a statistic, which follows an asymptotic normal distribution. We repeated the procedure to generate 100,000 simulated meta-analysis sets. Given the significance level ╬▒ = 0.05, the power was the proportion of sample sets whose meta-analysis p-value was Ōēż╬▒.
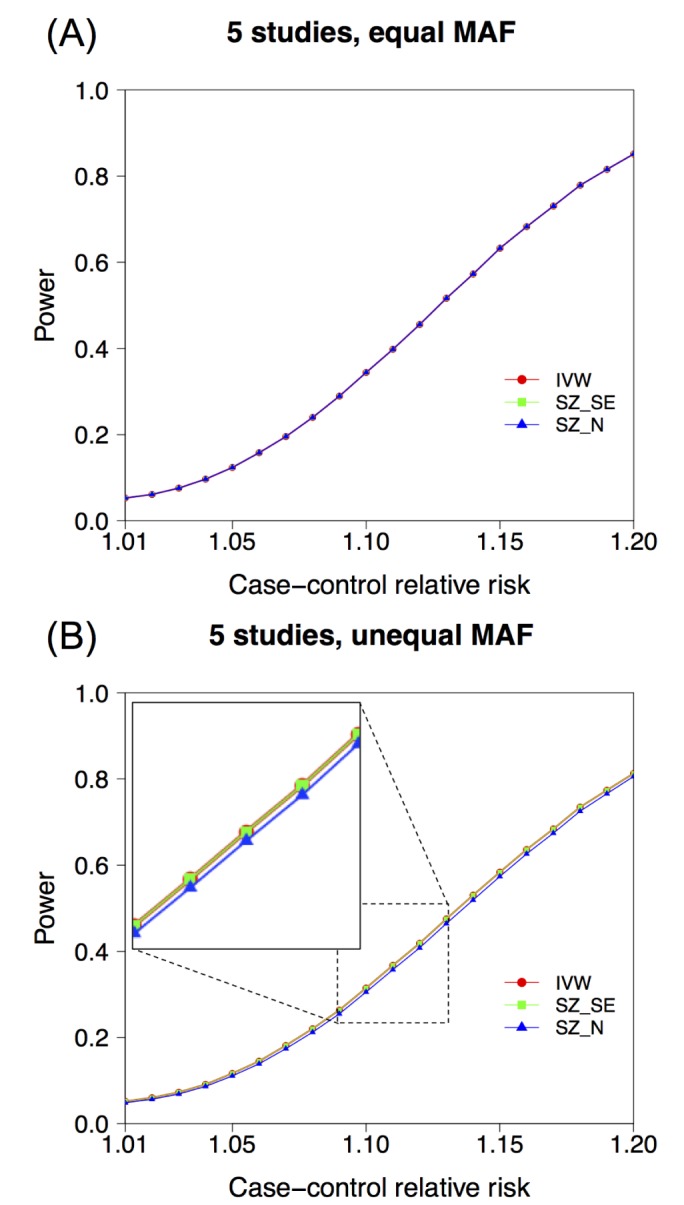
, ranging from 1.01 to 1.2. We assumed a meta-analysis of 5 genetic association studies and assumed a minor allele frequency (MAF) of 0.3. In an additional simulation setting, we assumed varying MAFs of (0.1,0.2,0.3,0.4,0.5) for the 5 studies. We assumed a very small disease prevalence (F Ōēł 0). Given these assumptions and parameters, we can calculate the expected MAF in cases and in controls. Specifically, given MAF p and relative risk ╬│, the case MAF becomes ╬│p/((╬│ŌłÆ1)p + 1), where the control MAF becomes approximately p, given F Ōēł 0. Given the expected MAF in cases and controls, we could randomly sample genotype data, assuming 500 cases and 500 controls for each of the five studies. To assess the statistical significance of the sample data, we used log odds ratio as a statistic, which follows an asymptotic normal distribution. We repeated the procedure to generate 100,000 simulated meta-analysis sets. Given the significance level ╬▒ = 0.05, the power was the proportion of sample sets whose meta-analysis p-value was Ōēż╬▒.We compared the two methodsŌĆöIVW and weighted SZŌĆöusing the inverse standard error as a weight factor (SZ_SE). Fig. 1 shows that the two methods showed the same power in both situations: under no heterogeneity in MAF between studies (Fig. 1A) and under heterogeneity in MAF (Fig. 1B). This result complements our analytical results that the two methods are equivalent if SZ uses SE(Xi)ŌłÆ1 as weights.
Additionally, we compared the two methods with another method, the weighted SZ, which uses the inverse squared root sample size as the weight (SZ_N). SZ_N was equivalent to IVW and SZ_SE in terms of power, when there was no heterogeneity in MAF (Fig. 1A). However, with the existence of differences in allele frequencies (therefore, differences in weights, wIVW,i ŌēĀ wSZ_N,i), SZ_N showed a slight power loss from the other two methods (Fig. 1B). This result demonstrates that using only sample size as the weight can be suboptimal if there are other factors that can cause variance differences between studies, such as allele frequencies. Nevertheless, the power drop of using only sample size as the weight was quite small (i.e., at ╬│ = 1.15, the power of IVW and SZ_SE was 58.24%, but the power of SZ_N was 57.23%, with only 1.01% power loss.)
Situations in which IVW and SZ can give distinct results
In this section, we demonstrate a situation in which IVW and SZ can give distinct results. As we have shown above, SZ whose weights are given as SE(Xi)ŌłÆ1(SZ_SE) is analytically equivalent to IVW. However, SZ whose weights are given as the square root of sample size (SZ_N) can give slightly different results, if the expected relationship S E X i ŌłÆ 1 ŌłØ N  is broken. We have already shown that a MAF difference can result in such breakage of this relationship. Here, additionally, we show that the use of a linear mixed model can also result in such breakage of the relationship
is broken. We have already shown that a MAF difference can result in such breakage of this relationship. Here, additionally, we show that the use of a linear mixed model can also result in such breakage of the relationship S E X i ŌłÆ 1 ŌłØ N S E X i ŌłÆ 1 ŌłØ N S E X i ŌłÆ 1 ŌłØ N
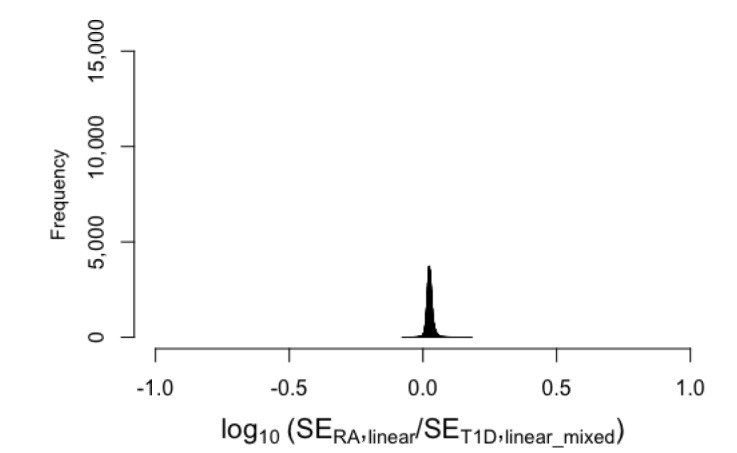
is broken. We have already shown that a MAF difference can result in such breakage of this relationship. Here, additionally, we show that the use of a linear mixed model can also result in such breakage of the relationship . We used the data of the Wellcome Trust Case Control Consortium [28]. This dataset includes approximately 2,000 cases for each of seven different diseases and 1,500 controls for each of two control groups (1958C and National Bureau of Standards [NBS]). We used the data on rheumatoid arthritis (RA) and type 1 diabetes (T1D). After standard quality-control and removal of the MHC region, we obtained 469,225 SNPs. We performed association tests for two diseases; we performed association tests for RA using NBS as controls and association tests for T1D using 1958C as controls. The sample sizes for the two association tests were similar (N = 3,318 and 3,443, respectively). We used logistic regression implemented in plink (with--logistic command). Because the sample sizes of the two tests were similar, we expected that for each SNP, the standard errors of the effect size estimate would be similar. That is, we wanted to test if the relationship held well for these real data. Fig. 2A shows that when we plotted the log10 value of the ratio of the two standard errors, the values were highly concentrated around 0. This implies that the standard errors were very similar between RA and T1D, as expected, because the sample sizes were similar. Thus, the relationship approximately held well. Therefore, Fig. 2A shows that IVW and SZ_N have similar results in this situation.Next, we changed the study design and used the linear mixed model implemented in the software package Genome-wide Efficient Mixed Model Association (GEMMA). We used GEMMA for both T1D and RA. Fig. 2B shows that the standard errors of these two analyses were similar. Then, we used GEMMA for T1D but not for RA. When we plotted the log10 value of the ratio of the resulting standard errors, the values deviated dramatically from 0 (Fig. 2C). The standard errors were much smaller in the linear mixed model than in the logistic regression. This is expected, because the effect sizes of the linear mixed model and logistic model have different meanings and are not comparable. Therefore, the relationship S E X i ŌłÆ 1 ŌłØ N
does not hold. As a result, in the meta-analysis, the p-values of IVW and SZ_N differed dramatically. Note that the standard errors given by GEMMA can be slightly different from the standard linear model, because GEMMA regresses the effect of population structure. However, an additional analysis comparing the standard errors of GEMMA and the standard linear model demonstrated that this effect is minimal and that their standard errors were similar (Fig. 3). Thus, the difference observed between GEMMA and the logistic regression model was mainly due to the use of different models (linear and logistic).One may argue that a meta-analysis design that combines the results of a logistic regression model and linear model is uncommon. Indeed, for binary traits, the use of a logistic regression model is more suitable. However, for dealing with population structure and cryptic relatedness, a linear mixed model is currently the main tool. For GWASs, there is no widely used efficient package implementing a logistic mixed model. For this reason, many studies are using a linear mixed model for binary traits as approximations. Therefore, if we assume a situation that the effect size in one study is obtained from a logistic regression model and the effect size in another study is obtained from a linear mixed model, the results of IVW and SZ_N can be different.
Discussion
In this paper, we investigated the optimal characteristics of two fixed effects meta-analysis methods: the inverse variance-weighted average and the weighted SZ. We showed that the two methods are optimized with different goals, but they are equivalent under certain assumptions. By analytical derivations and empirical simulations, we demonstrated their equivalency and provided insights into the optimal weights for the weighted SZ.
We have also shown that the optimal weights for the weighted SZ can be a function of not only the sample size but also other propertiesŌĆöfor example, allele frequencies in GWASs. We empirically showed that if allele frequencies differ between studies, using only sample size for the weight can be suboptimal in terms of power. Therefore, we suggest that one should use effect size and standard error to define a z-score and use the inverse of the standard error as the weight for the weighted SZ. The standard error term includes all information, such as sample size and allele frequencies, thus providing optimal performance. Nevertheless, in our simulations, using just sample size resulted in only slightly lower power (at most, 1.07% power loss). Thus, in most applications, using only sample size for weights might perform reasonably well.
We also demonstrated that in some situations, the two meta-analysis methods can give different results. Specifically, when one study used the linear mixed model to account for population structure, the effect size from the linear mixed model can be incompatible with the effect size from the logistic regression model. In such situations, the use of meta-analysis methods based on z-scores or p-values is recommended, because it is not sensible to apply inverse variance-weighted average to multiple incompatible effect sizes from different statistical models.