


Introduction
The concept of epistasis, generally defined as interactions among different genes, was first introduced in 1909 by William Bateson to describe the latent effect of one locus over another locus. A quantitative definition to the interaction was proposed in 1918 by R.A. Fisher as a statistical deviation from the additive effects of two loci on a phenotype. This definition enabled interaction analyses by testing whether products of multiple genotypes are statistically associated with phenotypes. More definitions about the gene-gene interaction have been proposed, but some are still not clearly understood. The statistical gene-gene interaction has often been confused with a biological gene-gene interaction. Particularly, the inference on a biological mechanism is complicated because of the lack of direct correspondence between statistical and biological interactions [1]. In general, statisticians define a statistical interaction as a departure from additivity in a linear model using a selected measurement scale [2]. However, as was pointed by Wang et al. [3], if one aims to infer biological interactions, statistically modeled interactions and main effect terms should not be interpreted separately [2]. In this paper, we detected gene-gene interactions with a likelihood ratio test. Genotype scores for single-nucleotide polymorphism (SNP) pairs were considered nominal variables, and nine different levels were assumed for a full model. For a reduced model, we considered three levels for each SNP, and thus, our likelihood ratio tests followed a chi-square distribution with 4 degrees of freedom. Therefore, the proposed method can detect biological interactions. It should be noted that biological interactions include statistical interactions.
The method of detecting gene-gene interactions has attracted much attention in genome-wide association studies (GWASs). Including logistic regression analysis for detecting gene-gene interactions, new methods, like comparing linkage disequilibrium (LD) in case and control groups, have been recently proposed [4]. However, analyzing a large number of SNPs in a GWAS is computationally very intensive, and various approaches, such as MDR [5], BEAM [6], Random Jungle [7], PLINK [8], and BOolean Operation-based Screening and Testing (BOOST), have been proposed to enable gene-gene interactions on a genome-wide scale.
In this paper, we considered BOOST method, proposed by Wan et al. [9]. BOOST uses a non-iteratively estimated measure that is approximately equal to the maximum likelihood estimators for a log-linear model, and it is used to select pairs of SNPs with a specified threshold. There is an updated method, graphical processing units BOOST (GBOOST), which is a BOOST method implemented for a graphical processing units framework for enabling parallel computing to achieve massive assignment in a fast manner [10]. GBOOST achieves a 40-fold speedup compared with BOOST. However, in the algorithm of BOOST, covariates other than SNPs can not be considered. We need a more flexible approach to improve the power of the model.
Here, we propose an efficient strategy that combines the BOOST screening stage and logistic regression method. Logistic regression generally shows good statistical power for a wide spectrum of epistasis. Screening with BOOST is a computationally efficient screening method, and a genome-wide search can be completed within a few hours. A follow-up stage of logistic regression with covariates would improve the statistical power of the model. In this paper, we first review the BOOST method and apply the proposed two-stage approach to type 2 diabetes (T2D) in a Korean population. This analysis of gene-gene interactions on a genome-wide scale with BOOST was completed within 42 hours, and we also identified several pairs of SNPs associated with T2D.
T2D is the most common form of diabetes, and unlike people with type 1 diabetes, T2D patients make insulin. However, either their pancreas does not make enough insulin or the body cannot use the insulin well enough. The prevalence of T2D has increased rapidly in recent years. The prevalence of T2D in Korea was estimated to be 7.3% (in people over 20 years of age) in 2005, and the rate of patients with T2D is expected to increase dramatically from 7.08% in 2010 to about 10.85% by 2030 [11]. Even more and more children are being diagnosed with T2D. Environmental effects, like obesity and lack of physical activity, are two of the most common causes of T2D, and the increasing prevalence may be related with them. It is also known that T2D has considerable heritability, which indicates a genetic effect. Until now, genetic variants in nearly 70 loci have been identified for T2D. Some variants from genes, such as KCNQ1 [12], KCNJ11, PPARG, NRF1, IDE, TCF7L2, CDKAL1, HHEX, IGF2BP2, CDKN2A/B, and SLC30A8 [13], were reported as significant susceptible genes to T2D in the East Asian population. To detect the relation between gene-gene interactions and T2D phenotype, we performed a gene-gene interaction analysis with the proposed two-stage analysis. We analyzed 8,842 participants (4,183 males and 4,659 females) with 352,228 autosomal SNPs collected from the Korea Association Resource project (KARE). We found some promising gene-gene interactions with the proposed method, and they will be investigated further in our future follow-up studies.
Methods
The KARE cohort
The KARE project started in 2007 and recruited 10,000 participants aged between 49 to 60 years from Ansung and Ansan, in the Gyeonggi Province of South Korea. About 50 million autosomal SNPs were genotyped with the Affymetrix Genome-Wide Human SNP arrary 5.0 [14]. In total, 8,842 individuals with 352,228 SNPs are available. In our GWASs, we discarded SNPs for which the Hardy-Weinberg equilibrium p-values were less than 10ŌĆō5, the genotype calling rates were less than 95%, and the minor allele frequencies were less than 0.05. We also eliminated subjects with gender inconsistencies, those whose identity by state was more than 0.8, and those whose calling rates were less than 95%. As a result, we analyzed 8,773 participants (4,117 males and 4,656 females) with 304,245 SNPs.
Definition of T2D
An individual was coded as a T2D patient if the condition satisfied the World Health Organization (WHO) diabetes diagnostic criteria: fasting plasma glucose (glu0) Ōēź 126 mg/dL, plasma glucose (glu120) Ōēź 200 mg/dL 2 h after an oral dose, or glycated hemoglobin (HbA1c) Ōēź 6.5%. A total of 1,169 subjects were diagnosed as cases, and the other individuals were considered controls.
Notations
We assume that there are L SNPs and n subjects. Genotypes at SNP l are denoted by Xl, where l = 1, ŌĆ”, L, and Y indicates the disease status: 1 for case and 2 for control. We assume that SNPs are bi-allelic, and capital and lowercase letters always indicate the major and minor alleles, respectively. For instance, AA indicates a homozygous reference genotype, Aa indicates the heterozygous genotype, and aa indicates the homozygous variant genotype. For simplicity, we denote the homozygous reference genotype, heterozygous genotype, and homozygous variant genotype as 1, 2, and 3, respectively.
Review of BOOST
The logistic regression model with only a main effect for two SNPs, p and q, can be modeled by the following form: and denote its log-likelihood as LM. The logistic regression model with main effects and interaction terms is
We denote it log-likelihood value by LF. Then, the interaction effects can be detected by the difference of the maximum log-likelihoods (MLEs) of these two modelsŌĆöi.e., L ^ F - L ^ M  .
.
.However, the difference in the log-likelihood needs very intensive computation for hundreds of billions of pairs of SNPs. Alternatively, there exists a one-to-one correspondence between a logistic regression model and a log-linear model in categorical data analysis [15], and BOOST considers interaction models based on log-linear models [9].
On the basis of the equivalence between the log-linear model and its corresponding logistic regression model, BOOST constructed its test statistic using the homogeneous association model MH and saturated model MS and denotes their log-likelihood as LH and LS, respectively. Then, we denote the observed genotype count of disease status k with Xp = i and Xq = j by nijk and the expected genotype count by ┬Ąijk, where k = 1 or 2, i = 1, 2, or 3, and j = 1, 2, or 3. Then, the maximum log-likelihood of both models will be
If we let ╬╝ ^ i j k H  be the MLE of ┬Ąijk for MH, we have
be the MLE of ┬Ąijk for MH, we have
be the MLE of ┬Ąijk for MH, we have
The interaction effects based on the likelihood ratio test can be calculated by the following forms:
If we let n be the total sample size, ŽĆ ^ i j k = n i j k n 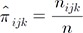 and
and P ^ i j k = ╬╝ ^ i j k H n  , it can be further simplified as and it can also be denoted by Kullback-Leibler's [9] form as
, it can be further simplified as and it can also be denoted by Kullback-Leibler's [9] form as
and , it can be further simplified as and it can also be denoted by Kullback-Leibler's [9] form as
This provides another interpretation of interactions, in that the difference of two log-likelihoods is proportional to the Kullback-Leibler divergence of the joint distribution obtained under the saturated model MS and the distribution obtained under homogeneous association model MH.
In particular, there is no closed form solution for homogenous association model MH, and iterative methods are needed to calculate the likelihood ratio tests. Likelihood ratio tests are computationally intensive when facing hundreds of billions of SNP pairs in the interaction analysis.
To address this issue, BOOST uses Kirkwood superposition approximation (KSA) [16] to estimate under MH as
Therefore, it can be utilized to approximate LH. If we let LKSA be the likelihood based on KSA, then we get following equation: and it can be calculated easily on the basis of the contingency table. Through simulation analysis by Wan et al. [9], 2 L ^ S - L ^ K S A  is known to be an upper bound of
is known to be an upper bound of 2 L ^ S - L ^ H  , and they are almost identical if they are larger than 25. The default value of the threshold Žä in BOOST is 30, and its corresponding p-value is 4.89 ├Ś 10ŌłÆ6 [9].
, and they are almost identical if they are larger than 25. The default value of the threshold Žä in BOOST is 30, and its corresponding p-value is 4.89 ├Ś 10ŌłÆ6 [9].
is known to be an upper bound of , and they are almost identical if they are larger than 25. The default value of the threshold Žä in BOOST is 30, and its corresponding p-value is 4.89 ├Ś 10ŌłÆ6 [9].In the screening stage of BOOST, all pairwise interactions will be evaluated by using KSA. If 2 L ^ S - L ^ K S A
>Žä, the interaction will be considered in the next testing stage; otherwise, it will be discarded from the analyses. All of the non-significant SNP pairs would be filtered out in this manner in the screening stage.Two-stage gene-gene interaction analyses
We first apply the BOOST approach to select the promising pairs of SNPs with the a priori chosen Žä. Žä can be selected based on the available computing facility, and we set Žä = 30 for our analyses. After filtering SNP pairs from the first stage with BOOST, we apply the logistic regression. BOOST can not adjust for the effects of covariates, and we applied logistic regression analysis with adjustments for sex, age, body mass index (BMI), and the top 10 principal component (PC) scores to the selected pairs of SNPs with BOOST. The logistic regression analysis was performed by using the glm function in R software. To calculate the p-values of the interaction term, we used the ANOVA function by comparing two fitted models in R.
Results
We have carried out an interaction analysis on T2D in the KARE cohort on the genome-wide scale; 8,773 subjects with 304,245 SNPs were considered for detecting gene-gene interactions. Missing genotypes were imputed with Impute2 [17] software.
We applied the proposed two-stage approach to identify the genome-wide significant gene-gene interactions. The analyses were completed within 42 h with an Intel Core i3-4130 CPU 3.40 GHz desktop. A total of 46,282,357,890 interactions were executed, and the Bonferroni-adjusted 0.05 genome-wide significance level is 1.08e-12. Promising pairs of SNPs were selected with BOOST, and 229,965 pairs of SNPs were selected with BOOST; then 229,965 pairs of SNPs were analyzed with logistic regression.
To perform adjustments for the population substructure between individuals, we used the EIGENSTRAT [18] method. EIGENSTRAT calculates the genetic similarities among subjects by using a genetic relationship matrix and applies PC analysis. The generated PC scores are then utilized as covariates for genetic association analyses, and this approach guarantees robustness against a population substructure. Here, we calculated the first 10 PC scores, and they were included as covariates for the logistic method in R. Sex, age, and BMI were also included as covariates in the analysis.
Most significant results for interaction analyses with
KARE datasets are listed in Table 1. Only the top 10 significant SNP pairs are listed. The most significant interaction effect with a p-value of 3.11E-11 was found for rs1402142 and rs8012584. The former is associated with the genes HTRA3, AREG, TEC, and NRAS (Table 2), which are related to metabolic, immune, and hematological diseases, and the latter is associated with the genes ANG and ABCB1, related to neurological and metabolic diseases. Our results show that even the interactions are significant, but their marginal effects may not be. There are some interesting results that rs872234 is near DHFRP2, which is related to diabetes mellitus type 1 [19], and rs1110144 and rs1104853 are both in CNTNAP2 [20] and MIR548T, respectively; they are also known to be associated with diabetes.
Discussion
The analysis of gene-gene interactions on a genome-wide scale is computationally very intensive, and many computational and statistical approaches have been recently proposed to minimize the computational burden. We found that BOOST is highly computationally efficient and can filter out non-significant interaction pairs in a fast manner.
In this study, we proposed an efficient strategy to identify interactions in genome-wide SNP data. We first utilized the screening stage of BOOST to filter out non-significant pairs and then used logistic regression with several covariates, such as age, sex, BMI, and PC scores.
In real data analysis, we used the KARE cohort dataset to detect gene-gene interactions of T2D. The smallest p-value (3.11E-11) of interaction pairs in the KARE data was found for rs1402142 and rs8012584. The Bonferroni-adjusted genome-wide significance level is 1.08e-12, and this SNP pair is not significant genome-wide. This insignificance is partially attributable to the insufficient sample size. With advances in genotyping/sequencing technology, genotyping costs will be much lower, and therefore, in the near future, sufficiently large samples will become available for genegene interaction analyses, which may lead us to a better understanding of human diseases.




