Introduction
Human genome variation has facilitated the understanding of inter-individual phenotypic differences [1, 2]. In addition to single-nucleotide polymorphisms (SNPs), it is widely accepted that large-scale DNA structural variation, termed copy number variation (CNV), is a major component of human genetic diversity [2, 3]. Genomewide SNP genotyping data can be used for CNV calling [4]; therefore, if we can get reliable CNV calls from the SNP genotyping data, a CNV-based genomewide association study (GWAS) can be realized. Among many whole-genome CNV analysis platforms, SNP arrays have been suggested as a resource for CNV discovery due to their ubiquitous genome coverage and relatively advantageous resolution. However, despite the importance of CNV-disease association analysis, CNV calling from SNP genotyping data has not been well established. Affymetrix Genomewide SNP 5.0 is one of the commonly used SNP array platforms for SNP-GWAS as well as CNV analysis [5]. We previously validated the accuracy and reproducibility of CNVs called from Affymetrix SNP array 5.0 data by comparing the CNV calls from 3 different array platforms using NEXUS software: Affymetrix SNP array 5.0, Agilent 2X244K CNV array, and NimbleGen 2.1M CNV array [6].
Recently, a number of CNV defining algorithms have been developed, which have facilitated the CNV-based GWAS [7-14]. However, due to the fundamental limitation of SNP genotyping data for the measurement of signal intensity, there are still concerns regarding the possibility of false discovery or low sensitivity for detecting CNVs [15, 16]. Indeed, CNV calling is dependent on the types of array platforms and analytic tools. Each platform and calling algorithm has its own advantages and disadvantages; so, one single algorithm or array platform is not always best for determination of CNVs [17-19]. Recently Pinto et al. [20] showed that different analytic tools applied to the same raw data typically yielded CNV calls with <50% concordance and, using multiple algorithms, minimize the number of false discoveries. To remedy the potential limitations of SNP array for CNV detection, more than one way of CNV calling by using several different segmentation algorithms are performed, and overlapped calls are used for GWAS analysis [21-24].
In this study, we tried to verify the effect of adopting multiple CNV calling algorithms and set up the most reliable pipeline for CNV calling with Affymetrix Genomewide SNP 5.0 data. We selected the 3 most commonly used algorithms for CNV segmentation from SNP genotyping data, PennCNV, QuantiSNP, and BirdSuite. After defining the CNV loci using the 3 different algorithms, we assessed how many of them overlapped with each other, and we also validated the CNVs by genomic quantitative PCR (qPCR). Finally we concluded that CNVs that were consistently called from more than 2 different calling algorithms are more reliable than the CNVs called from a single algorithm. Our result will be helpful to set up the CNV analysis protocols for Affymetrix Genomewide SNP 5.0 genotyping data.
Methods
Study materials
We used Affymetrix Genomewide SNP 5.0 genotyping data provided by the Korea Association Resource (KARE) consortium, Korean Genome Epidemiology Study (KoGES). As a control, we purchased a HapMap cell line, GM10851, from Coriell Institute for Medical Research (Camden, NJ, USA) and extracted genomic DNA using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany).
Pre-processing SNP array data
Before CNV calling procedures, all required pre-processing procedures, including allele correction, summarization, and background correction, were performed as described previously by using the software provided by Affymetrix [5]. In brief, background-corrected data were normalized using quantile normalization and summarized by median polish.
CNV calling
For this study, Affymetrix 5.0 SNP array (Affymetrix, Santa Clara, CA, USA) data of 10 subjects were randomly selected from the KARE dataset for CNV calling. Affymetrix 5.0 data of NA10851 was used as a control. For defining CNVs, we choose 3 different segmentation algorithms, PennCNV [25], QuantiSNP [26] and BirdSuite [27]. PennCNV implements a hidden Markov model (HMM) and considers SNP allelic ratio distribution in addition to signal intensity. We only used CNVs from PennCNV with samples that had a standard deviation of log R ratio (LRR) smaller than 0.2, drift values of b-allele frequency (BAF_drift) smaller than 0.01, and waviness factor between -0.05 and 0.05. For QuantiSNP, we did not apply any criteria for sample filtering, since QuantiSNP uses an Objective Bayes Hidden-Markov Model (OB-HMM) for calibrating the model for fixing the false positive error rate and maximum marginal likelihood to set other hyperparameters. It was originally developed for detecting CNVs from BeadArray SNP genotyping data of Illumina, but Affymetrix data also could be processed as long as they have probe signal intensities and B-allele frequencies. Birdsuite uses exclusive copy number analysis routine (Canary) for CNP locus and HMM for finding rare CNVs. It generates a logarithm of the odds ratio (LOD) value for each CNV that describes the likelihood of a CNV relative to no CNV over a given interval, including flanking sequences. Of the BirdSuite calls, only the CNVs that have an LOD value smaller than 2 were used for further analysis. All 3 methods were used with default values, and no other option was added or changed.
Validation
To estimate the false discovery rate of our CNV-calling algorithm, CNVs were validated by genomic real-time qPCR. For this purpose, we randomly selected 25 CNV loci and performed genomic qPCR using DNAs from study subjects who showed corresponding CNVs on that loci. The PCR primers used in this study were designed using the PrimerQuest program (http://www.idtdna.com/Scitools/Applications/Primerquest). To verify the specificity of the PCR reactions under the unified denaturation temperature (60℃), we performed PCR and agarose gel electrophoresis for each primer set. We also screened the University of California Santa Cruz (UCSC) database (http://genome.ucsc.edu/) to confirm the unique sequence without any repeat sequences in the primers. Sequence information of the primers for genomic qPCR validation is listed in Table 1.
Ten microliters of the reaction mixture contained 10 ng of genomic DNA, SYBR Premix Ex Taq TM II (TaKaRaBio, Shiga, Japan), 1×ROX (Toyobo, Osaka, Japan), and 10 pmol of primers. Thermal cycling conditions consisted of 1 cycle of 10 s at 95℃, followed by 40 cycles of 5 s at 95℃, 10 s at 61℃, and 20 s at 72℃. All PCR experiments were repeated twice, and amplification efficiencies for both target and reference genes were evaluated using a standard curve over 1:5 serial dilutions. The copy number of each target was defined as 2-ΔΔCT, where ΔCt is the difference in threshold cycles for the sample in question normalized against the reference gene (RNaseP) and expressed relative to the value obtained by calibrator DNA (NA10851 and Promega DNA), as described elsewhere [28].
Results and Discussion
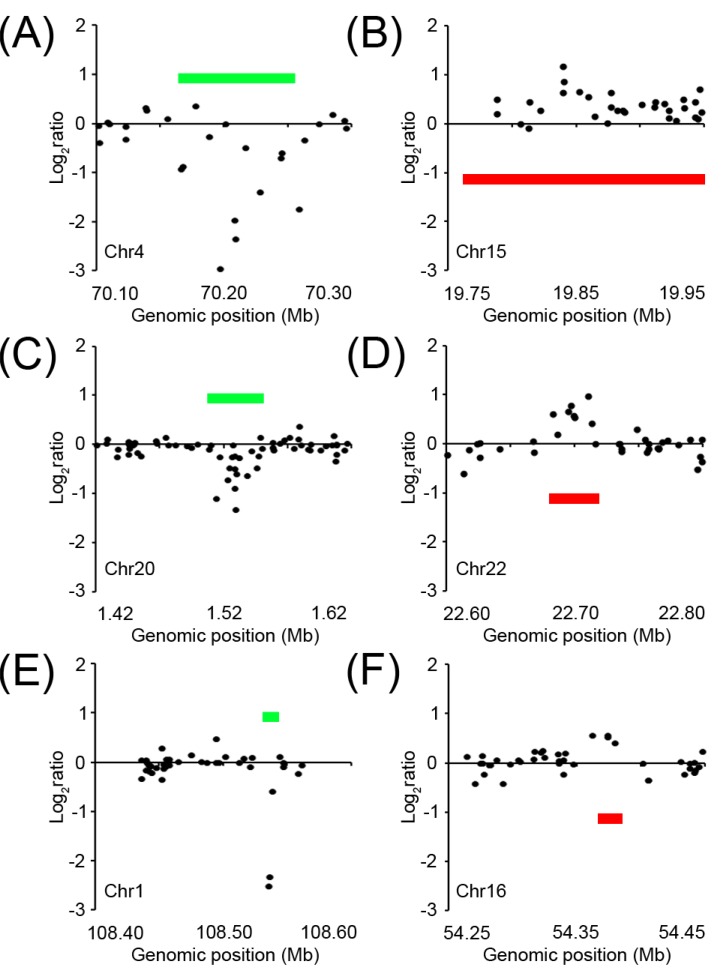
The general characteristics of the CNV calls from the 3 algorithms are shown in Table 2. The total number of CNV calls from the 3 algorithms was similar-range 814 to 1,362. The median size of the CNVs called by PennCNV (12.9 kb) were bigger than the other 2 algorithms (7.5 kb and 7.9 kb). The size of the CNVs consistently detected by 2 or more algorithms was generally bigger than that of the CNVs detected by a single method. For example, the average sizes of the CNVs that were detected from all 3 algorithms, 2 algorithms, and 1 method were 107 kb, 73.2 kb, and 15.9 kb, respectively (Table 3). Fig. 1 shows the examples of different sizes of CNVs according to the LRR distribution of gain- and loss-type CNV regions, depending on how many algorithms detected the CNV in that region. These data suggest that larger-sized CNVs are generally more prominent; so, they can be relatively easily detected by any segmentation method, regardless of the algorithm. But, detection of the smaller-sized CNVs can be affected more easily by the characteristics of the CNV defining algorithm; so, they can be detected by 1 particular algorithm.
A recent report examining the impact of inaccuracy of CNV detection from SNP genotyping data revealed that CNVs, defined as the copy number changes of >7 consecutive probes, were fairly reliable in case of deletion type CNVs [29]. In our previous study exploring the CNV profiles of Koreans using an Affymetrix array, we suggested the filtering condition of CNV call as >6 consecutive probes to be reliable [5]. Therefore, to make more reliable conditions in this study, we filtered the CNV calls as >7 consecutive probes. Under this criterion, the number of CNV calls from the 3 algorithms was reduced down from 295 to 385, while the median size became bigger-50 kb by PennCNV and QuantiSNP and 23.5 kb by BirdSuite (Table 2). In this condition, same as above, the size of the CNVs consistently detected by 2 or more algorithms was generally bigger than that of the CNVs detected by a single method (Table 3).
In terms of the number of CNVs consistently identified by different algorithms, of the CNV calls using the PennCNV algorithm, 48.6% (396/814 CNVs) was detected by 2 or more algorithms, and only 4.9% (40/814 CNVs) was detected by all 3 algorithms. Similarly, of the CNV calls using QuantiSNP, 42.9% (466/1,087 CNVs) was detected by 2 or more algorithms and only 3.9% (42/1,087 CNVs) was detected by all 3 algorithms. However, in the case of the BirdSuite algorithm, although the number of CNV calls was the biggest, only 9.6% (131/1,362 CNVs) was detected by 2 or more algorithms and 2.9% (40/1,362 CNVs) was detected by all 3 algorithms. Taken together, only 17% of the total CNV calls (465/2,735 CNVs) were defined by more than 2 algorithms, and only 1.5% (40/2,735 CNVs) was defined by all 3 algorithms (Table 3). In the filtering condition of >7 consecutive probes, the number of CNVs consistently identified by different algorithms was improved. For example, of the CNV calls using the PennCNV algorithm, 76.6% (226/295 CNVs) was detected by 2 or more algorithms and only 23.4% (69/295 CNVs) was detected uniquely by PennCNV. QuantiSNP calls showed a similar trend. However, even under this condition, 92.5% (356/385 CNVs) of BirdSuite calls were unique (Table 3). All the CNVs consistently detected by 2 algorithms were from PennCNV and QuantiSNP, while no CNVs that were detected by the BirdSuite algorithm were consistently detected by 2 algorithms, with only 1 exception. These data suggest that CNV calls from SNP genotyping depend substantially on the characteristics of calling algorithms, and only part of the CNV calls from each algorithm seems to be reliable. Therefore, CNVs identified from SNP genotyping data must be validated as completely as possible, and especially in the case of using a single calling algorithm, the CNVs must be validated more carefully. Filtering the CNV calls by the number of consecutive probes can improve the reliability of CNV calls.
To validate the CNVs identified from 3 different algorithms in this study, we randomly selected 25 CNV regions across the whole chromosomes. The genomic qPCR validation results are listed in Table 4. In case of the CNVs identified by all 3 algorithms, 71.4% (5/7) of the consistency was observed between the CNV call and genomic qPCR result. Of the CNVs identified by more than 2 algorithms, 57.6% (19/33) was consistent. Of the unique CNV calls defined by just a single algorithm, only 38.3% (18/47) was consistent, but the other 61.7% (28/46) was not consistent. In the case of the CNVs defined as >7 consecutive probes, the consistency was generally improved-47.8% (unique CNVs), 62.2% (CNVs identified by more than 2 algorithms), and 83.3% (CNVs identified by all 3 algorithms). These results indicate that CNV calls from 2 or more algorithms are more reliable than those from single algorithms. CNV calls from all 3 algorithms are, of course, the most reliable, but this is too stringent; so, the number of CNVs is not applicable for GWAS analysis.
In this study, we aimed to verify the effect of adopting multiple CNV calling algorithms and set up the most reliable pipeline for CNV calling with Affymetrix Genomewide SNP 5.0 data. We found that CNVs defined by a single CNV calling algorithm may not reliable enough for further GWAS study, regardless of the types of algorithms. Based on our findings, we propose that for reliable CNV-based GWAS using SNP array data, CNV calls must be performed with at least 3 different algorithms, and the CNVs consistently called from more than 2 methods must be used for association analysis.