Transposable Elements and Genome Size Variations in Plants
Article information
Abstract
Although the number of protein-coding genes is not highly variable between plant taxa, the DNA content in their genomes is highly variable, by as much as 2,056-fold from a 1C amount of 0.0648 pg to 132.5 pg. The mean 1C-value in plants is 2.4 pg, and genome size expansion/contraction is lineage-specific in plant taxonomy. Transposable element fractions in plant genomes are also variable, as low as ~3% in small genomes and as high as ~85% in large genomes, indicating that genome size is a linear function of transposable element content. Of the 2 classes of transposable elements, the dynamics of class 1 long terminal repeat (LTR) retrotransposons is a major contributor to the 1C value differences among plants. The activity of LTR retrotransposons is under the control of epigenetic suppressing mechanisms. Also, genome-purging mechanisms have been adopted to counter-balance the genome size amplification. With a wealth of information on whole-genome sequences in plant genomes, it was revealed that several genome-purging mechanisms have been employed, depending on plant taxa. Two genera, Lilium and Fritillaria, are known to have large genomes in angiosperms. There were twice times of concerted genome size evolutions in the family Liliaceae during the divergence of the current genera in Liliaceae. In addition to the LTR retrotransposons, non-LTR retrotransposons and satellite DNAs contributed to the huge genomes in the two genera by possible failure of genome counter-balancing mechanisms.
Introduction
Although some plants exist in aquatic environments, plants are mainly denizens of land. Since the first brave aquatic algae landed on plants in the Silurian period about 450 million years ago, plants had spread from the tundra to tropic and aquatic lake or ocean to alpines. The physical mass and size are also enormously variable, from only a few millimeters long in bryophytes to over 90 meters tall in eucalyptus. The sessile nature and lack of obvious immune systems of green plants may have adopted different life strategies compared to species in the animal kingdom.
The genome, the genetic material of an organism, has been adopted for ambient environments. As seen in the wide range of ecological niches and structural complexity, the cellular genome is also highly variable in plant species by as much as over 2,000-fold [1, 2]. Cellular DNA content has been measured in over 6,000 plant species [1, 2], and highquality DNA sequence information is available from about 50 plant species [3]. Prior to the genomic era, the general idea was that plant genomes constitute a large amount of 'junk' or 'parasitic' or 'selfish' DNAs [4, 5], which are mostly repetitive DNAs and transposable elements. However, these DNAs have been revisited with a plethora of genomic information and considered genomic gold in the evolution of eukaryotic taxa [6, 7, 8]. The extreme genome size variation of eukaryotes, especially in plant taxa, has posited a conundrum in evolutionary genetics. The recently updated information of whole-genome sequences is shedding light from diverse angles to understand genome size variations with evolutionary and biological significance in many organisms. This review excerpts topics on genome size variation in relation with transposable elements. In the last session, the transposable elements and genome evolution in the Liliaceae family, known to have large genomes in angiosperms, will be discussed.
Eukaryotic Genome Architecture
The redundant nature of eukaryotic genomes was analyzed well before the dawn of modern genomic analysis. Waring and Britten [9] demonstrated that not all DNA coded for genes in eukaryotes by reassociation kinetic analysis of mouse DNA. The DNA reassociation kinetic analysis, Cot analysis, allows one to dissect the genomic composition in several species, in which the genomes of complex organisms can be fractionated into several classes depending on the repetitiveness, such as unique, slightly repetitive, moderately repetitive, and highly repetitive sequences [10]. The Cot analysis was very useful in the analysis of the genome complexity in species with large genomes, such as major crop plants [11, 12, 13].
When the eukaryotic chromosomes are dissected at the molecular level, sequence composition is not even along the chromosomes. While the low-copy protein coding sequences are clustered in discrete blocks between various repetitive DNA sequences, the repeated DNA sequences are located at a few defined sites or widely dispersed, depending on the classes of repetitive DNA [14, 15]. The chromosomal landmarks of repeated sequences include centromeres, telomeres, nucleolus organizing region (NOR), and heterochromatin. While centromeres house highly repeated satellite DNA sequences and Ty3/gypsy long terminal repeat (LTR) retrotransposons, Ty1/copia LTR retrotransposons and microsatellites or simple sequence repeats are dispersed and present in clusters [15, 16]. The 18S-5.8S-25S rRNA gene repeats are on the NORs, which appear as secondary constrictions in cytological preparations [14, 15]. Another repeat of the 5S rRNA gene is also clustered in one or more sites in chromosomes [14, 15, 16]. The chromosomal tips, telomeres, also contain many thousands of the telomere repeat TTAGGG [14, 17]. Besides these repeat motifs, many other types of intercalary tandem repeats or dispersed repeats are also scattered along the chromosomes.
Eukaryotic nuclear DNA is packaged into chromatin structure. During interphase, the local structure of chromatin depends on the genes present in the DNA. The low-copy protein-coding genes are loosely packaged into euchromatin, whereas sequences of protein-noncoding genes are tightly packaged into the gene desert heterochromatin. The local chromatin structure can be altered by epigenetic chemical modification of the DNA and chromatin-associated proteins. The chromatin structure dynamic is a critical process in the regulation of DNA replication, gene expression control, and cell division [18]. Some points of the epigenetic control of gene expression and DNA replication are beyond the scope of this review; so, epigenetic chromatin modifications will be dealt here in detail only in relation with genome size and transposable elements.
Transposable Elements (TEs)
Since the discovery of transposable elements in maize in 1940s by McClintock [19, 20, 21, 22], TEs have been found to be ubiquitously present in eukaryotes [23]. TEs are capable of moving their locations within the genome so that they can generate genomic plasticity by inducing various chromosomal mutations and allelic diversity [24, 25, 26]. TEs are conventionally classified into two classes based on the transposition mode: class 1 TEs and class 2 TEs [27]. Class 1 TEs are the retrotransposons that retrotranspose via an mRNA intermediate in a "copy-and-paste" process that can lead to extremely high copy numbers in the genome. Class 2 TEs comprise transposons that transpose directly via DNA by a "cut-and-paste" process that allows them to attain moderate copies in the genome. With the plethora of information of whole-genome sequences in a growing number of species [3, 28], more and more families of TEs are being mined, which necessitates the adoption of a unified classification system for TEs. This systematic classification groups the TEs into 2 major classes, 9 orders, and 29 superfamilies by the mode of chromosomal integration, gene structure, and sequence similarity [23].
Class 1 TEs can be subdivided into several subclasses but generally into two subclasses: LTR retrotransposons and non-LTR retrotransposons. Although both LTR and non-LTR retrotransposons are in plant genomes, LTR retrotransposons are more prevalent than non-LTR retrotransposons in plant species. LTRs range from a few hundred base pairs to several kb (kilobases), and LTRs start with nucleotides of 5'-TG-3' and end with 5'-CA-3'. Because the LTR retrotransposons integrate into 4- to 6-bp staggered cut sites, they produce 4- to 6-bp target site duplication (TSD) upon integration. The LTR retrotransposons encode two open reading frames, GAG and POL [29, 30, 31]. GAG encodes a protein for replication, and POL encodes protease (PR), integrase (INT), reverse transcriptase (RT), and RNase H (RH). The LTR retrotransposons are divided into two superfamilies, Ty1-copia (Pseudovirideae) and Ty3-gypsy (Metavirideae), based on the order of genes they encode [23]. The overall sequences are similar, but the gene order is PR-INT-RT-RH in Ty1-copia and PR-RT-INT-RH in Ty3-gypsy retrotransposons, respectively. Phylogenetic analyses of the RT domain revealed a monophyletic origin of the Ty1-copia and Ty3-gypsy subfamilies [32, 33]. Moreover, the high sequence similarity and gene structure between Ty3-gypsy elements and retroviruses indicate that retroviruses evolved from Ty3-gypsy elements by acquisition of the env gene [31, 34].
Although non-LTR retrotransposons are less prevalent than LTR retrotransposons in plant taxa, four main orders of class 1 non-LTR retrotransposons, Dictyostelium intermediate repeat sequence, Penelope-like element), long interspersed nuclear elements and short interspersed nuclear element), are found in plants [23, 31]. DIRSs carry unusual split direct repeats or an inverted repeat at both ends and do not produce TSD upon integration. Termini of PLE elements are LTR-like, but their orientation can be either a direct or an inverted orientation. They have an RT and endonuclease domain, and the RT domain of PLE is more similar to telomerase than the RT of LTR retrotransposons. Both long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) lack LTRs at their termini. While LINEs can reach several kb, SINEs are short (80-500 bp). LINEs are predominant elements in animal genomes, whereas they are not prevalent in plant genomes except the Del-2 element, which is highly abundant in the genomes of species in the genus Lilium. The Del-2 elements will be discussed in more detail in the following session. Of the five major superfamilies (R2, L1, RTE, I, and Jokey) in the LINE order, only L1 and I are present in plant genomes [23, 31]. SINEs are also highly abundant in animal genomes, especially in mammalian genomes; they are relatively less dominant in plant genomes. The best known SINE is the Alu element, which is present in up to 500,000 copies in the human genome [35]. The origin of SINEs is obscure and is presumed to have originated from various polymerase III transcripts. Their AT-rich 3'-region carries 35-bp tandem repeats or contains a poly(T) tail, the pol III termination signal [35].
Class II DNA transposons can be subdivided into subclass 1 of terminal inverted repeat (TIR) transposons and subclass 2 of non-TIR transposons [23, 31]. The subclass 1 TIR DNA transposons are flanked by various lengths of TIRs and produce various lengths of small TSDs upon integration. The TIR transposons encode a transposase, which excises the element from the genome and integrates into a new site in the genome, because it has both endonuclease and ligase activity [36, 37, 38]. The TIRs form a secondary structure where transposases bind. Most of the known transposases carry a DDE catalytic motif, which comes in close proximity to each other and forms a catalytic center to enable the enzyme to excise the transposon DNA from the original site and reinsert it into the target site [39]. The subclass 1 TIR DNA transposons are classified into 10 superfamilies based on the sequence similarities. Of the 10 superfamilies, plant genomes harbor 6 superfamilies: Tc-Mariner, hAT, Mutator, P, PIF-Harbinger, and CACTA.
In the subclass 2 non-TIR DNA transposons, two non-TIR DNA transposon superfamilies, Helitrons and Maverick, are present. Helitrons are widespread, diverse, and abundant in some plants, but their identification is difficult due to their lack of a terminal repeat structure [40]. The Helitrons replicate via a rolling-circle mechanism and do not produce TSDs upon integration. Helitrons can attain very high copy numbers and create genomic diversity. Indeed, they are present in high copies, and captured gene fragments of pseudogenes contribute to maize genome diversity [41, 42, 43].
Depending on the movability both class 1 and class 2 TEs can be further classified into autonomous and non-autonomous elements. While the autonomous elements equip all the machinery in mobilizing themselves, the non-autonomous elements lack one or all of the components of this machinery [44]. The non-autonomous elements are decayed autonomous elements by deletion or other mutations, but they are highly abundant in plant genomes [23]. Transposition of class 1 elements is a multi-step process. That is, they must be transcribed and translated to produce proteins to reverse-transcribe the mRNA and package it into virus-like particles. The non-autonomous class 1 retrotransposons lack self-encoded proteins, so that the multi-step is blocked. However, they can be mobilized if the missing proteins are provided in trans [45]. The BARE1 and BARE2 elements illustrate an example of the autonomy and non-autonomy of the class 1 retrotransposons. BARE1 (Barley RetroElement1) is a copia-like retrotransposon and highly abundant in the barley genome [46]. It is an autonomous element, and the protein machinery encoded is produced. The BARE2 is a non-autonomous element, having most of its protein-coding domains intact, except for the defective Gag gene. The defect of BARE2 was complemented by the BARE1 machinery to retrotranspose in the barley genome [47]. In class 2 DNA transposons, the non-autonomous elements cannot express transposase to catalyze their own transposition. Because they were derived from autonomous elements, they form a binary system in terms of mobilization, which was recognized in the Ac/Ds system by the early pioneering observation in maize by McClintock [22]. miniature inverted repeat transposable elements) are worthwhile mentioning here among the non-autonomous class 1 transposons [48, 49, 50]. Because MITEs are small and carry only the minimal feature of TIR, they have been elusive in the identification of autonomous elements to mobilize them to attain very high copies in plant genomes. By an extensive bioinformatics search, it was revealed that Stowaway MITEs were derived from the Tc1/mariner superfamily, and Tourist MITEs were derived from the PIF/Harbinger superfamily [51]. Thus, it was deduced that MITEs can burst by autonomous elements in these superfamilies.
C-values of Spermatophyte
C-value or genome size refers to the total amount of DNA in an unreplicated haploid or gametic nucleus of an organism [52]. C-value varies tremendously by as much as 660,000-fold among eukaryotes, from 2.3 Mb in Encephalitozoon intestinalis, a parasitic microsporidian, to 148,880 Mb in Paris japonica, a monocotyledonous plant in Liliales [53, 54, 55]. The general trend of C-value is that simple organisms have small genomes and complex higher organisms have large genomes. However, the C-value variation is even present among closely related species, and genomic DNA content is often poorly correlated with organismal complexity, which projects the C-value paradox [56].
Plant taxa show more C-value variation than other taxa (Table 1) [3, 57]. Among land plants, the genome size variation is as much as a 2,348-fold difference from the smallest, Genlisea margaretae (1C = 63.4 Mbp), to the largest, Paris japonica (1C = 148,880 Mb). Large size differences exist even in a single genus at the same ploidy level as in the genus Eleocharis, which comprises more than 250 species, in which E. acicularis (2n = 20, C = 0.25 pg) is 20 times smaller than E. palustris (2n = 16, C = 5.5 pg) [58]. Using Feulgen microdensitometry or flow cytometry, C-value has been measured in over 6,000 plant species and categorized in the Kew Plant DNA C-value Databases (http://data.kew.org/cvalues/) [1, 2]. The average genome size (1C) is 5.809 Gbp (1 pg = 980 Mbp) in angiosperms and 18.157 Gbp in gymnosperms, respectively. The median 1C-value is 2.401 Gbp for angiosperms and 17.506 Gbp for gymnosperms, respectively; indicating that genome size in angiosperms is not evenly distributed but skewed, favoring a small size.
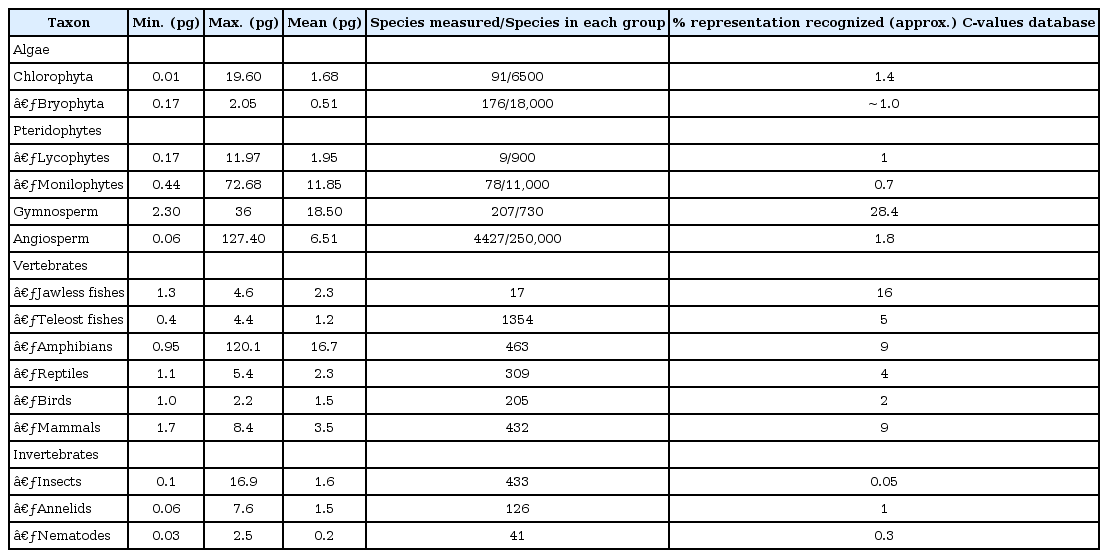
Minimum, maximum, and mean 1C amounts in several major groups of plants and animals
With the rapid development of high-throughput sequencing technologies, the list of complete genomes that have been sequenced is growing sharply to provide deep insights into genomic evolution by comparing whole-genome information [28, 59, 60]. In plants, genome sequences are available in more than 50 species, including 36 dicots, 16 monocots, 1 gymnosperm, and 1 each of a lycophyte and bryophyte [28]. The genome size in the list ranges from the smallest genome of the carnivorous Utricularia gibba (bladderwort) at 82 Mb [61] to the largest genome of a gymnosperm species, Picea abies (Norway spruce), at 19,800 Mbp [62]. The most frequent genome size of the sequenced plant genomes is about 500 Mbp, which is similar to the average genome size in the Kew Plant DNA C-value Database (http://data.kew.org/cvalues).
Transposable Elements and Genome Size
Repeated DNA sequences are predominant in the plant genome. The repeat sequences range from 3% of the small genome of U. gibba [61] to 85% of the large genome of maize [63]. Transposable elements constitute most of the repeated DNAs. Of the two classes of transposable elements, class 1 retrotransposons constitute significant proportions in plant genomes. Of the first 50 plant genomes sequenced, the number of full-length LTR retrotransposons is available in 21 species, in which the genome size increment correlated with the abundance of repeated sequences (r2 = 0.584) and LTR retrotransposons (r2 = 0.68) [28].
The "cut-and-paste" class 2 DNA transposons multiply their copies when the transpositions occurs from a recently replicated genomic region into unreplicated genomic regions or when the excision site is repaired by gene conversion using the intact sister chromatid as a template [64]. Thus, the class 2 DNA transposons do not usually attain high copy numbers, and their impact on genome size is minimal. Among class 2 elements, MITEs are an exception in copy number, because they are present at extremely high copy numbers in some genomes [65, 66]. However, the small size of MITEs, 200-500 bp, has a limitation on their impact on genomic obesity [67].
The semiconservative "copy-and-paste" transposition can increase copy numbers of the class 1 retrotransposons continuously. Indeed, the magnitude of C-value variation in plant genomes cannot be explained solely by whole genome duplication (WGD), and genome contents are highly correlated with the amount of class 1 retrotransposons among the sequenced genomes [28, 67]. Unless there are mechanisms of removing these class 1 retrotransposons or other repeated sequences, plants may end up with a 'one-way ticket' to genomic obesity [68]. Does a large genome actually cause any problem in the adaptation of host species? In a sense, the dramatic increase of noncoding or repeated sequences must give some burden to the cell for replication and metabolism, which was postulated by the 'large genome constraint hypothesis' [69]. That is, the nutritional requirements of the extra phosphorus (P), a major constituent of DNA, and subsequent longer replication times may result in longer generation times and a reduction in fecundity, leading to selective disadvantage [70]. Actually, plants having a 1C-value of less than 3 pg could be annual, biennial, or perennial, whereas plants having a 1C-value of larger than 25 pg cannot grow quickly enough to be annual and are to be obligate perennials [71]. Genome sizes of angiosperms are skewed, favoring a small size, except for a few extreme large genomes [1, 67]. The fact that lower unicellular organisms usually have relatively small genomes implies that genomic obesity by redundant sequences must have posed a negative impact on their survival. Nevertheless, some plant genomes have survived with so many burdensome large fractions of repeated DNAs, implying that the negative effect of redundant DNAs may have been negated by some compensation. Of the 50 sequenced plant genomes, the median C-value was about 500 Mbp. The species with around 500 Mbp genomes (rice, sorghum, etc.) have genomes that are about 30% transposable elements; so, it was argued that the average transposable element fraction is around 30% of the plant genomes [28]. Large genome information with high accuracy and effective bioinformatics tools have unveiled the biological roles of repeated DNAs, for which several excellent reviews are available on this aspect [6, 72, 73, 74].
The ever increasing class 1 retrotransposons can be counterbalanced by genome-purging mechanisms that include illegitimate or unequal recombination between LTRs and other types of deletions [75]. Intrastrand homologous recombination between directly repeated LTRs deletes the sequences between LTRs, leaving solo LTRs [76]. Analysis of solo LTRs and comparison of internally deleted retrotransposons showed that illegitimate intrastrand homologous recombination may be the driving force to maintain slim genomes in Arabidopsis [77] and rice [78]. Recently inserted TEs are often removed from the genome, leading to rapid genomic turnover. For example, the rice genome experienced several bursts of LTR retrotransposons during the last 5 million years but ultimately removed over half of the inserted LTR retrotransposon DNA [79]. The cotton genus, Gossypium, provided another example genome expansion and contraction. The diploid Gossypium species showed 3-fold differences in genome size, from 88 Mbp in the D-genome of New World cotton to 2,572 Mbp in the K-genome of Australian cotton [80]. Gorge3 is a gypsy-like retrotransposon in the Gossypium species. The expansion and contraction of Gorge 3 were lineage-specific phylogenetically among Gossypium species. The smaller genomes showed not only slow gain of Gorge3 compared to the larger genomes but also effective removal of Gorge3 elements, which exceeded the rate of the gain [76].
Non-homologous end joining (NHEJ) after double-strand break (DSB) can also lead to massive genomic restructuring by purging LTR retrotransposons in a small genome [81]. The genome of Oryza brachyantha is 60% smaller than its close relative cultivating species, Oryza sativa, in which 50% of the size difference was due to the amplification and deletion of recent LTR retrotransposons. Of the 32,038 protein-coding genes of O. brachyantha, only 70% of them were in collinear positions in comparison with the rice genome. So, the authors argued that the low LTR retrotransposon activity and massive internal deletions of the LTRs by NHEJ after DSB shaped the current O. brachyantha genome. Similarly, NHEJ after DSB was found to be a common pathway in the genomic reduction of A. thaliana [82].
The genome of Norway spruce (Picea abies) sheds another interesting view on genomic evolution in gymnosperms. It is the only genome sequenced in gymnosperms [62] and the largest genome among the first 50 sequenced plant genomes [28]. The sequence data showed no evidence of WGD since the divergence with angiosperms 350 million years ago. Surprisingly, LTR retrotransposons of the Norway spruce genome are mostly low-copy LTRs with 80% singletons. The large genome seems to have resulted from a slow but steady accumulation of diverse LTR retrotransposons, which was possible due to the lack of efficient elimination mechanisms, as evidenced by the fact that other pine species also have low-copy LTRs [80]. The 24-nucleotide small RNAs are involved in TE silencing [83], but expression of these small RNAs in Norway spruce is also low. The average gymnosperm genomes are relatively larger than those of angiosperms. But, the gymnosperms have not gone awry to obese genomes, as seen in some angiosperm species. More gymnosperm genome sequences will give some answers for this modest modulation of LTR retrotransposon families and genomic expansion in gymnosperms.
Arabidopsis lyrata revealed several interesting phenomena on genomic contraction and expansion. It is an out-crossing relative to the self-pollinating model species A. thaliana. The A. lyrata genome is 1.6 times larger than that of A. thaliana [84]. Comparison of the two genomes revealed that there were large-scale genome rearrangements, but most differences in genome size were derived from hundreds of thousands of small deletions in A. thaliana, mostly in non-coding areas and retrotransposons [84]. A subsequent comparative study with other plant genomes showed that A. thaliana lost introns 6 times faster than A. lyrata [77]. In a genome-wide survey of synonymous substitution rates, A. thaliana had a higher mutation rate than A. lyrata, which might relate to the higher rate of intron loss and rapid genome reduction [85]. In the mutational hazard hypothesis [86], non-coding DNA is prone to accumulate deleterious mutations and be purged. So, with the high mutation rate in A. thaliana and intron loss, the DNA loss process may still be ongoing in A. thaliana genomes, suggesting pervasive selection for smaller genomes [84].
Epigenetics and Genome Size
Transposon insertions and copy number increases are potentially detrimental to the host genome. As a consequence, eukaryotic genomes employ epigenetic surveillance systems that allow TEs to be under-controlled. Ironically, however, Nina Fedoroff proposed a somewhat different view on plant genome size variations with transposable elements in her seminal AAAS presidential address: "I argue that transposable elements accumulate in eukaryotic genomes because of, not despite, epigenetic silencing mechanisms" [26]. Nevertheless, epigenetic regulations of the transposable elements are the first line of defense against uncontrolled transposable element proliferation [75]. Epigenetic mechanisms regulate gene expression and do not involve changes in DNA sequence. They include DNA methylation [83, 87, 88], chromatin remodeling [89], and siRNAs [88, 89]. By genomic, epigenomic, and population genetic analyses with A. thaliana, Hollister and Gaut [83] presented three results: i) gene expression is negatively correlated with the density of methylated TEs, ii) methylated TEs near genes are under purifying selection, but unmethylated TEs or TEs far from genes are not under purifying selection, and iii) TE insertions are distributed by age and methylation status, so that older, methylated TEs are farther from genes. Thus, host silencing of TEs near genes has deleterious effects on neighboring gene expression, resulting in the preferential loss of methylated TEs from gene-rich euchromatic regions. The young LTRs near genes are targeted for DNA methylation by guidance of 24-nt small interfering RNA, which results in the silencing of LTRs as well as silencing of expression of nearby genes [83, 88]. The comparison of A. thaliana with its LTR-laden and larger relative A. lyrata can back up this view, because LTRs in A. thaliana are further from genes by more effective purging than those of A. lyrata [84, 88]. So, it is an evolutionary trade-off in which beneficial TE silencing casts a fitness cost by the deleterious effects on the expression of nearby genes [83].
In maize, the mop1 mutant has a deficiency of RNA-dependent RNA polymerase, which is a component of the RNA-directed DNA methylation pathway [89]. In a global gene expression analysis of the mop1 mutant, most of the class 2 DNA transposons (78%) showed increased expression, but 68% of the analyzed class 1 retrotransposons showed decreased expression [90]. Several histone deacetylases, enzymes involved in heterochromatin formation, increased their expression in the mop1 mutant, implying increased heterochromatin formation in the mutant. Thus, there are several pathways that lead to TE silencing, and these pathways interact with each other, so that some families are rigorously suppressed but expression of some families is relaxed, which permits a rapid burst of some transposable families in a short period of time [84, 91].
Genome Size Evolution in the Genera Lilium and Fritillaria
The genomes of the species in Liliaceae are highly variable in 1C-value, ranging from 3.4 pg in Prosartes smithii to 127.4 pg in Fritillaria assyriaca [92]. Especially, the species in the subclasses Lilieae and Tulipeae in the Lilioideae subfamily harbor large genomes in the Liliaceae family. By comparative analysis of the relationship of genome size with phylogenetic among 78 species in Liliaceae, Leitch et al. [92] demonstrated that the ancestral genome size of the Liliaceae species was 6.73 pg, and punctuated genome size evolution occurred twice in Liliaceae. The first genome size evolution was at the base of the polytomy divergence in Liliaceae, which was associated with a shift from the smaller genomes to the larger genomes of the Lilioideae (median C value, 14.2-50.9 pg) and Medeoloideae (median C value, 14.2-18.9 pg) subfamilies. Table 2 shows the genomic features of two genera, Lilium and Fritillaria, in the Lilioideae subfamily. The second genomic evolution was at the divergence of Lilioideae from Medeoloideae, which was about 12 million years ago [93]. Punctuated genomic evolution that led to speciation was also observed at the molecular level in other species [94]. The subfamily Medeoloideae is a sister subfamily to Lilioideae in the Liliaceae family. While the species in Medeoloideae bear rhizomes, inconspicuous flowers, fleshy animal-dispersed fruits, and broad reticulate-veined leaves, the species in Lilioideae show bulbs, showy flowers, winddispersed seeds, and narrow parallel-veined leaves in open habitats [93, 95]. Because these ecological shifts and the adaptive morphological features coincided with the appearance of a large genome size, an adaptive role of genome size in Lilioideae is suggested-i.e., if the large genome has any selective advantage. However, as argued by Leitch et al. [92], there was no tendency toward increasing genome size from the ancestral species, having about 6.73 pg. Their proposal was that the large genomes of Lilioideae were a necessary or random consequence, simply due to the large diversification at the base of the clade. Oliver et al. [96] proposed a theory that genomic size evolution is proportional to genome size, with the fastest rates occurring in the larger genomes, among 20 major eukaryotic clades. So, the larger genomes of Lilium and Fritillaria in Lilioideae might have drifted away to even larger genomes with an increased rate of genome size evolution [92].
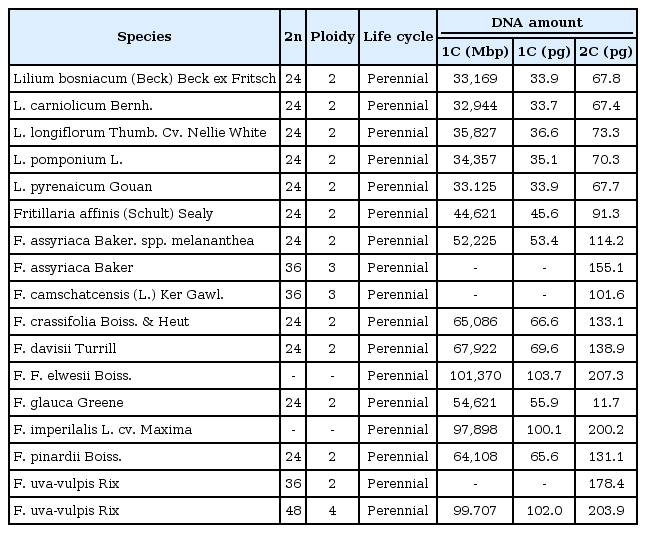
Genomic features of the species in the genera Lilium and Fritillaria
Although species in the genus Lilium have been known to carry large genomes, studies of transposable elements have not been carried out extensively. The genome of the species in the genus Lilium spans from 32.8 to 47.9 pg, with a mean value of 39.6 pg [92]. A Ty3/gypsy retrotransposon, del-1, was isolated from Lilium henryi [86]. Del-1 Ty3/gypsy was present in approximately 13,000 copies in the L. henryi genome, which accounts for 1% of the genome of L. henryi. By DNA reassociation analysis, Joseph et al. [97] showed that Cot1 repeated DNAs constitute 2.2% of the L. henryi genome, whereas they constitute 9.7% in L. longiflorum. In a Southern blot analysis with 14 Lilium species, del-1 Ty3/gypsy retrotransposons showed high copy variation, at least a 100-fold difference, among Lilium species, implying that the del-1 Ty3/gypsy element was highly active after divergence of the genus Lilium. The integration time of del-1 Ty3/gypsy was estimated to be about 0.6 million years ago (mya) in L. henryi and 1.6 mya in L. longiflorum [16]. Del-2 is a LINE-like element found in Lilium speciosum [98, 99]. Del-2 is 4.5 kb in size and is present in 240,000 copies, constituting about 4% of the L. speciosum genome [99]. Del-2-related sequences were also highly abundant in Tulip and bluebells (Scilla nonsripta, both in Liliaceae), Hyacinthaceae, Amarylidaceae, and Alliaceae plants but less abundant in other monocots, such as Poaceae, implying that they are specific to the Liliaceae family. LINEs are highly abundant in mammals, but they are scant in most plant species [23, 31]. However, the del-2 LINE-like elements are highly abundant in species of the Liliaceae family. LIREs (lily retroelement) are Ty1/copia retrotransposons isolated from L. longiflorum [16]. LIREs showed approximately the same abundance as the del-1 Ty3/gypsy retrotransposon in species of the genus Lilium. However, unlike the del2 LINEs, LIRE Ty1/copia retrotransposons are absent in other species, such as tulips, Narcissus, Ipheion, Hosta longgips, and O. sativa. We mined eight Ty1/copia elements from Lilium lancifolium (unpulished). Characterization of these retrotransposons may provide more information on the Ty1/copia retrotransposons in the genus Lilium. The genus Fritillaria is a sister genus in the Lilieae subclass in the Lilioideae subfamily. The Fritillaria species harbor an even larger genome (min. 30.8 pg, max. 89.2 pg, mean 50.9 pg) than Lilium species [92]. In F. affinis (1C = 45.6 pg) and F. imperialis (1C = 43.0 pg), chromovirus-like sequences and Tat lineage Ty3/gypsy retrotransposons are predominant in both species [100]. Not only do retrotransposons contribute to the gigantic Fritillaria genome, satellite DNAs also FriSAT1 repeats also contribute highly to the Fritillaria genome. The FriSAT1 repeats constitute approximately 26% of the F. affinis genome [100]. With these results, they argued that there are no predominant repeats that track the increasing/decreasing trends of genome size in Fritillaria. Instead, many diverse families of transposable elements and repeats constitute the gigantic Fritillaria genomes. Thus, as evidenced with full-genome sequencing of the large genome of Norway spruce (P. abies) [80, 82], the highly obese Fritillaria genome may have had happened partly by the failure of the repeats to be purged effectively to counterbalance the amplification of retrotransposons.
Conclusion
Large fractions of eukaryotic genomes contain repeated DNA sequences that are mostly transposable elements. The large quantity of accurate genomic information in various plant taxa and the development of computational biology have revolutionized our conventional view on transposable elements, which were once dishonorably christened 'selfish DNAs' and have now been switched to 'genome treasure.' Genome size variation is enormously variable in plant taxa, and most of this variation is due to the variable amounts of class 1 LTR retrotransposons in the host genomes. The 'copy-and-paste' retrotransposition of the class 1 LTR retrotransposons left the plant genomes to get into genomic obesity. The uncontrollable amplification of class 1 LTR retrotransposons could have caused a negative effect on host genomes not only by the various mutational effects but also by the large genomic constraint in replication. Thus, epigenetic surveillance systems control the activity of retrotransposons and counterbalance genome size amplification by genome-purging mechanisms, such as illegitimate or unequal recombination between LTRs and NHEJ. The epigenetic surveillance systems, however, might have been weakened in certain times, and some retrotransposons in certain lineages had escaped the epigenetic radar to have an amplification burst in a relatively short period of time during plant evolution and diversification. However, this specific amplification might have not happened in all plant taxa, as seen in the Norway spruce, which has a relatively larger genome than average angiosperm genomes. Although we need more gymnosperm sequence information, the steady accumulation of diverse LTR retrotransposons with weakly efficient elimination mechanisms can also account for the genome sizes in gymnosperms. In addition to LTR retrotransposon amplification, as seen in Lilium and Fritillaria, non-LTR retrotransposons and simple satellite DNAs also contributed to the large genomes that happened partly by the failure of effective purging to counterbalance the growth of genome size. Biological (hybridization, alien chromatin introgression, environmental stresses, or pathogens, etc) and nonbiological (temperature shift, UV irradiation, etc.) stresses might have caused the weakening of epigenetic surveillance to allow genome bursts in certain species. So, the current genomes may be the outcome of a long battle of repeat sequence amplification and genome-purging systems in ambient environments during plant evolution.
Acknowledgments
This work was funded by a Korea Research Foundation grant (Grant No. NRF-2012R1A1A2043773) and a grant from the Golden Seed Project, Ministry of Agriculture, Food and Rural Affairs (MAFRA), Ministry of Oceans and Fisheries (MOF), Rural Development of Korea (RDA), and Korea Forest Service (KFS).