New Lung Cancer Panel for High-Throughput Targeted Resequencing
Article information
Abstract
We present a new next-generation sequencing-based method to identify somatic mutations of lung cancer. It is a comprehensive mutation profiling protocol to detect somatic mutations in 30 genes found frequently in lung adenocarcinoma. The total length of the target regions is 107 kb, and a capture assay was designed to cover 99% of it. This method exhibited about 97% mean coverage at 30× sequencing depth and 42% average specificity when sequencing of more than 3.25 Gb was carried out for the normal sample. We discovered 513 variations from targeted exome sequencing of lung cancer cells, which is 3.9-fold higher than in the normal sample. The variations in cancer cells included previously reported somatic mutations in the COSMIC database, such as variations in TP53, KRAS, and STK11 of sample H-23 and in EGFR of sample H-1650, especially with more than 1,000× coverage. Among the somatic mutations, up to 91% of single nucleotide polymorphisms from the two cancer samples were validated by DNA microarray-based genotyping. Our results demonstrated the feasibility of high-throughput mutation profiling with lung adenocarcinoma samples, and the profiling method can be used as a robust and effective protocol for somatic variant screening.
Introduction
Non-small-cell lung cancer is an increasingly common and lethal disease, accounting for 25% of all cancer deaths. Sequencing of the lung cancer genome is of particular interest for identifying driver mutations and their pathways involved in cancer growth and development [1]. Somatic mutational profiles are crucial for cancer diagnosis and classification, which lead to tailoring the best therapeutic strategy to individual patients [2].
Previous studies using the Sanger sequencing method have identified several key mutations associated with lung cancer. Massive PCR amplification and Sanger sequencing of 623 candidate cancer genes in 188 lung adenocarcinomas discovered 26 mutational target genes [3]. Although the study provided highly valuable results, it is very time-consuming and costly-so much so that a single laboratory can hardly perform this kind of large-scale sequencing projects.
A mass spectrometric-based mutation detection technology, named OncoMap, has been effective in identifying somatic mutations in cancer genomes [4]. Currently, it can detect more than 1,000 mutations in 112 commonly mutated genes that were previously identified as oncogenes and tumor suppressors [5]. Although OncoMap is a high-throughput method for mutational profiling with both fresh frozen and paraffin-embedded tissue samples, the mutation detection is limited to previously identified mutations, and it cannot discover novel mutations.
Recent advancements of next-generation sequencing technology have made breakthroughs in identifying unknown somatic mutations [6]. Combined with sequencing technology, targeted enrichment techniques have been developed to reduce sequencing cost and time [7]. Several recent studies have reported targeted resequencing of cancer samples using next-generation sequencing technologies [8, 9].
Here, we present a fast and cost-effective method to identify somatic mutations in lung cancer. We surveyed the literature and chose 30 mutational target genes that were associated strongly with lung adenocarcinoma development. Target gene capture was performed using selector probes, which involved circularization and amplification of specific restriction fragments covering the target genes using rolling circle amplification [10]. The captured target DNAs were analyzed by next-generation sequencing to find somatic variations. This method could be useful to detect previously known recurrent mutations as well as novel variations.
Methods
Preparation of genomic DNA from cancer cell lines
We ordered normal genomic DNA sample from the Coriell Institute Cell Repository (CCR ID NA17022; Camden, NJ, USA), which originated from a normal Caucasian male of European descent. For cancer genomic DNAs, H-1650 and H-23 cancer cells were cultured and harvested for DNA preparation. Genomic DNA (gDNA) was extracted with the QIAamp DNA blood kit according to the manufacturer's instructions (Qiagen, Hilden, Germany). The DNA quality and quantity were assessed with the use of a Nanodrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA).
Design and oligonucleotides
The list of the entire exons for the 30 genes was obtained from a consensus coding sequence database, CCDS (build 36.3), showing a total number of 701 exons covering 102 kb, according to hg18 (March, 2006 assembly).
Targeted restriction fragments were selected using Disperse software [11]. Templates for circularization of each chosen targeted fragment (selector probes) were designed using ProbeMaker software [12]. Each selector probe consisted of two sequences of 20-25 nucleotides complementary to the ends of its targeted restriction fragment.
The 3'-biotin-labeled oligonucleotides (Integrated DNA Technologies, Coralville, IA, USA) were prepared by incubating the oligonucleotides with 1× Tdt buffer (NEB), 1× CoCl2 (NEB), 0.1 mM dUTP-biotin (Roche Diagnostics, Mannheim, Germany), and 0.2 units/µL terminal transferase (NEB) in a final volume of 50 µL. The reaction was incubated at 37℃ for 1 h and followed by enzyme inactivation at 75℃ for 20 min.
Target enrichment
Eight different restriction reactions were used to digest genomic DNAs from each sample, including SfcI and Hpy188I in NEB buffer 4; DdeI and AluI in NEB buffer 2; MseI and Bsu36I in NEB buffer 3; MslI and BfaI in NEB buffer 4; HpyCH4III and Bsp1286 in NEB buffer 4; SfcI and NlaIII in NEB buffer 4; MseI and HpyCH4III in NEB buffer 4; and HpyCH4V and EcoO109I in NEB buffer 4 (New England Biolabs, Ipswich, MA, USA). The restriction reactions contained 1 unit each of two restriction enzymes and their corresponding compatible NEB buffer in 1× concentration and 0.85 µg/µL bovine serum albumin (BSA) in a total volume of 10 µL. The reactions were incubated at 37℃ for 60 min, followed by enzyme inactivation at 80℃ for 20 min.
A total of 80 µL of pooled digested sample was mixed with 10 pM biotinylated selector probes, 1 M NaCl, 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, and 0.1% Tween-20 in a total volume of 160 µL. The mixture was incubated and hybridized at 95℃ for 10 min, 75℃ for 30 min, 68℃ for 30 min, 55℃ for 30 min, and 46℃ for 10 h. The hybridized solution was mixed with 10 µL M-280 streptavidin-coated magnetic beads (3.35 × 107 beads/mL; Invitrogen, Carlsbad, CA, USA) in 1 M NaCl, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 0.1% Tween-20 in a final volume of 200 µL and incubated at room temperature for 10 min. After incubation, the beads were collected using a ring magnet and washed in 1 M NaCl, 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, and 0.1% Tween-20 in a total volume of 200 µL at 46℃ for 30 min with rotation.
Multiple displacement amplification
Genomic fragments were circularized by incubating the beads with 1× Ampligase reaction buffer, 0.25 U/µL Ampligase (Epicentre, Madison, WI, USA), and 0.1 µg/µL BSA in a total volume of 50 µL at 55℃ for 10 min. The circularized molecules were separated from the beads by incubation with 5 µL sample buffer at 95℃ for 10 min and collected with a ring magnet rack. The supernatant was incubated with 5 µL reaction buffer and 0.2 µL enzyme mix (Templiphi; GE Life Sciences, Piscataway, NJ, USA) at 30℃ for 4 h, followed by inactivation at 65℃ for 10 min.
Real-time quantitative PCR analysis
The enriched samples were analyzed with real-time quantitative PCR (qPCR) and DNA quantification to evaluate enrichment bias and specificity. PCR primers were placed randomly in the targeted regions and quality controlled using standard genomic DNA. The qPCR results were used to estimate how much target DNA was present in the amplification products, and then, the specificity (proportion target material) was estimated by measuring the amount of DNA in the reactions.
The enriched and pre-enriched control DNAs were diluted (final dilution 1:3,600) in a PCR mix containing 1× PCR buffer, 2 mM MgCl2, 1 unit Platinum Taq, 0.2 mM dNTP (10297-018; Invitrogen), 1× SYBR Green I, 10% DMSO, and 0.16 µM of either on-target primer or off-target primer to a total volume of 30 µL. The genomic reference DNA was 10 ng of template in the same PCR mixture as described above. The qPCR was performed using an LC480 Real-Time PCR system (Roche), and the conditions were as follows: 95℃ for 5 min followed by 40 cycles of (95℃ 15 s, 56℃ 30 s), with end-point measurement of the fluorescence after each completed cycle.
Library preparation and massive parallel sequencing
Libraries were prepared according to the manufacturer's instructions (Illumina, San Diego, CA, USA). Briefly, 5 µg of gDNA in 200 µL nuclease-free water was fragmented by a Bioruptor (Diagenode, Liege, Belgium) at high power for 30 min (30 s ON and 30 s OFF). Overhangs of fragmented gDNA were converted to blunt ends using T4 DNA ligase and Klenow enzyme. Subsequently, an 'A' base was added to the ends of double-stranded DNA using Klenow exo- (3' to 5' exo minus). The paired-end adaptor (Illumina) with a single 'T' base overhang at the 3' end was ligated to the products above. The PE adaptor-ligated products were separated on a 2% agarose gel and excised from the gel from approximately 400 bp to 500 bp. The sequencing libraries were bar-coded to allow sequencing of 6 samples in one lane of a flow cell. Size-selected DNA fragments were enriched by PCR with PE primers 1.1 and 2.1 (Illumina). The concentration of the libraries was measured on both a Nanodrop (Thermo Fisher Scientific) and Qubit IT (Invitrogen). Finally, the libraries were validated by a Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The gDNA library was sequenced using the Illumina genome analyzer GAIIx according to the manufacturer's instructions.
Short read alignment and variation calling
A total of 76-bp paired-end or single-end sequence reads with ~200-bp insert size were aligned to the human reference genome (NCBI build 37, hg19) with BWA algorithm1 ver. 0.5.8c and default parameters [13]. Two mismatches were permitted in a 45-bp seed sequence. Putative single nucleotide polymorphisms (SNPs) and small INDELs were called by the pileup vcf function of Samtools (ver.0.1.9) [14]. The options used in filtering variations were a minimum of 6 for mapping depth and quality threshold of 50 for single nucleotide variations (SNVs) and 20 for small INDELs. Predicted SNVs were compared with NCBI dbSNP version 131 (http://www.ncbi.nlm.nih.gov/projects/SNP/) to remove known SNP information. Non-synonymous SNV information was extracted by comparing UCSC (http://genome.ucsc.edu/) reference gene information with the somatic mutation list in the COSMIC cancer information database (http://www.sanger.ac.uk/genetics/CGP/cosmic/). PolyPhen (polymorphism phenotyping) was utilized to predict possible changes in protein structure and function resulting from a non-synonymous amino acid change.
Genome-wide SNP analysis
SNP genotyping was performed using the Axiom genotyping solution, including an Axiom Genome-Wide ASI 1 Array Plate and reagent kit, according to the manufacturer's protocol (Affymetrix, Santa Clara, CA, USA). Briefly, total genomic DNA (200 ng) was treated with 20 µL of denaturation buffer and 40 µL neutralization buffer, followed by amplification for 23 h using 320 µL of Axiom amplification mix. Amplified DNA was randomly fragmented into 25 to 125 base pair (bp) sizes with 57 µL of Axiom fragmentation mix at 37℃ for 30 min, followed by DNA precipitation for DNA clean-up and recovery. DNA pellets were dried and resuspended with 80 µL of hybridization master mix; 3 µL of suspended sample was kept for sample qualification. A hybridization-ready sample was denatured using a PCR machine at 95℃ for 20 min and 48℃ for 3 min. Denatured DNA was transferred to a hybridization tray and loaded onto a GeneTitan MC with an Axiom ASI array plate (Affymetrix). Hybridization continued on the GeneTitan for 24 h, followed by loading of ligation, staining, and stabilization reagent trays into the instrument. After chip scanning, the cel intensity file was normalized, and genotype calling was done using Genotyping Console 4.1 with Axiom GT1 algorithms according to the manufacturer's manual. The cut-off values for data quality control were DISHQC ≥ 0.82 for hybridization and QC call rate ≥ 97%.
Results
Capture design
The 30 genes known to be mutated in lung cancer were chosen for targeted resequencing (Table 1). The 701 coding regions in 30 genes covered 102 kb according to the consensus coding sequence (CCDS) database. Capture sequences to achieve redundant coverage over the coding regions were chosen based on length (100-1,000 bp) and GC content (20-65%) and to avoid repetitive genomic elements in the ends. After analysis of in silico-digested restriction fragments of the target regions, the best combinations of restriction enzymes were selected to provide over 99% coverage of targeted bases.

30× coverage of individual target genes after deep sequencing
Target enrichment analysis
A normal sample (NA17022) and two lung adenocarcinoma cell lines (H-1650 and H-23) were examined in this study. It is reported in the COSMIC database that the H-1650 cell line has a deletion mutation in the EGFR gene, while the H-23 cell line has mutations in the KRAS, STK11, and TP53 genes.
After target gene enrichment, the enriched DNAs were analyzed using qPCR with primers targeting the regions of interest, along with qPCR primers targeting irrelevant, non-amplified loci outside the target regions. The correlation between the replicates was high (average r2 = 0.97), and the majority of primer pairs clustered within a range of 3 Cts (threshold cycle in qPCR). Therefore, the enrichment was highly reproducible, and the enrichment bias was minimal.
The enriched DNAs showed an average Ct of 17 with the target primers, while primer pairs targeting loci outside of the target regions had average Cts of 33. This indicated that the enrichment was target-specific. The average specificity in the normal samples was 28% when calculated from the Ct and amount of enriched product (Table 2).

Target capture specificity analyzed by qPCR
Sequencing analysis of enriched DNAs
To analyze the enriched samples, we sequenced the DNAs using a GAIIx next-generation sequencing instrument (Illumina) and evaluated important metrics to consider actual coverage, specificity, and reproducibility across the targeted loci.
On average, 4.3 gigabases (Gb) was produced per sample, and they were mapped to the reference genome (NCBI build 37, hg19) at a 73-90% mapping rate (Table 3).

Mapping statistics of next-generation sequencing experiments
The 26-41% of the uniquely mapped reads were found in the region of interest, demonstrating moderate specificity of this approach. The normal sample showed the lowest mapping rate (73.03%) but the highest specificity (41.58%), indicating that cancer genomes are less efficient for exome sequencing due to genomic changes.
In addition, about 97% of the targeted bases were covered at more than 30× (Fig. 1). This high depth coverage could allow us to examine low-purity cancer samples, which are not normally analyzed by Sanger sequencing or genotyping tools. The actual coverage of the normal sample differed, depending on the gene. The coverage of most target genes was more than 95% at 30×, but two genes, CDKN2A and LTK, showed low coverage of 75% and 84%, respectively (Table 1).
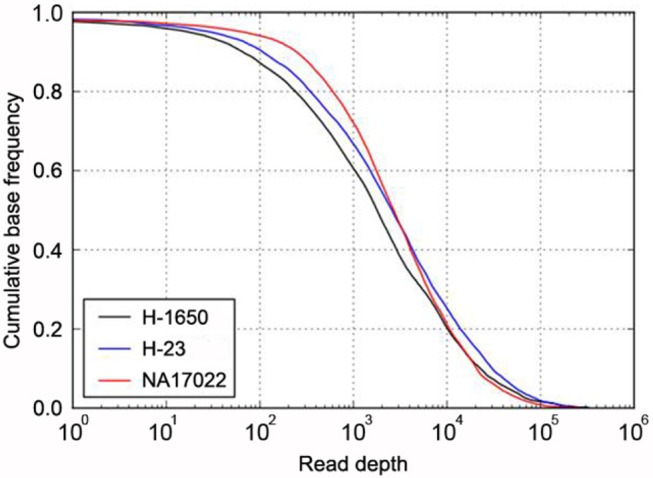
Target coverage in cancer and normal samples. The cumulative coverage of targeted bases (i.e., the fraction of all sequenced bases in the target regions that share more than a particular read depth) were plotted after sequencing 4.34 Gb of H-1650 (black), 6.03 Gb of H-23 (blue), and 3.25 Gb of NA17022 (red). The sequencing yield in the three samples resulted in 30× coverage of 92% (H-1650), 95% (H-23), and 97% (NA17022) of all target regions.
Using the final mapped reads, we constructed a genomic profile database for detecting SNVs (Supplementary Table 1) and short insertions and deletions (INDELs) (Supplementary Table 2). In each sample, we identified 81-179 SNVs and 50-90 small INDELs in target gene regions (Table 4). Cancer samples (H-1650 and H-23) showed about twice as many SNVs than the normal sample (NA17022).

Somatic variation candidates from the target gene regions
By subtracting SNVs found in the SNP databases, we identified cancer-specific somatic mutation candidates, and identical somatic mutations of the cancer cell lines in the COSMIC database were found. All previously known somatic mutations in the TP53, KRAS, and STK11 genes of sample H-23 and in the EGFR gene of sample H-1650 were identified in this study (Table 4).
The validity of the data was also examined with a genome-wide SNP microarray, which has 37 SNPs in the target region (Axiom Array; Affymetrix). The genotyping data showed 80-91% concordance without any bias (Table 5). The disconcordant variations were not biased to any sample, coverage, or genotype.

Comparison of SNV calls with DNA microarray genotyping results
Discussion
Our targeted resequencing method for somatic mutation profiling in lung cancer from 30 cancer-related genes produced unbiased target DNAs repeatedly. Analysis of the enriched DNAs by next-generation sequencing identified previously known mutations in the samples. Further analysis of more samples by targeted resequencing will reveal many novel variant candidates.
Target enrichment was performed using Selector technology (Halo Genomics, Uppsala, Sweden), which showed improved coverage and compatibility with next-generation sequencing library construction by employing rolling-circle amplification [10]. This method exhibited about 97% mean coverage at 30× depth, average 42% specificity, and high reproducibility (r2 = 0.98) of target enrichment in the normal sample (Tables 1, 2, 3), indicating that this is applicable to targeted resequencing of clinical samples. Although the enrichment specificity was moderate, this was overcome by increasing sequencing depth. As the total DNA bases of the target regions was 107 kb, 97% coverage at 30× depth was achieved by 3.25 Gb of sequencing, which does not create any cost issues by using next-generation sequencing technologies.
Exome resequencing has proven to be robust and effective for somatic variant detection in coding regions [31, 32]. Comparisons of sequencing data from normal and cancer tissues from individual patients have unveiled individual somatic mutation profiles. However, the cancer cell lines that we used had no normal cell pairs. To solve this problem, we tried to remove previously known normal variations as much as possible. Predicted SNVs from the sequencing data were further filtered using common variation information from the most updated dbSNP database. As a result, we found many somatic mutations, which included previously reported mutations in the COSMIC database, such as variations in TP53, KRAS, and STK11 of sample H-23 and in EGFR of sample H-1650 (Table 4). Especially, the number of reads that covered the four variations was more than 1,000. This provides many advantages when compared to whole-genome sequencing or whole-exome sequencing. Sequencing of cancer samples has raised several issues, such as sample purity and cancer heterogeneity. These shortcomings can only be overcome by in-depth sequencing of target regions. Therefore, our protocol is also useful for cost-effective somatic mutation screening of admixed clinical cancer samples.
Compared to whole-genome sequencing, targeted sequencing has an issue with uneven coverage of targeted genes. In Table 3, cancer samples showed fewer sequencing reads at target regions. This low target capture efficiency in cancer samples could be explained by genetic variations in cancer genomes that inhibit the hybridization between cancer DNA fragments and the designed capture oligonucleotides. This may be overcome by trial-and-error screening in selecting more efficient oligonucleotides.
There are two kinds of target capture technologies: hybridization and PCR. Hybridization-based target capture has been widely used and is able to cover more target regions but is time-consuming and hard to handle with many samples. In contrast, the PCR-based method is faster and allows us to handle more samples. Our protocol is a multiple displacement amplification-based method that is as efficient as the PCR-based method. For example, hybridization-based target capture methods, such as the Agilent SureSelect target enrichment kit and NimbleGen SeqCap EZ kit, normally handle 1-8 samples at the same time, while our protocol with a liquid handler could process 96 samples in parallel. This costs 12 times less money. Therefore, our target enrichment is scalable and easy to handle with multiple samples. This can result in remarkable reduction of total cost when combined with multiplexed next-generation sequencing. Therefore, our target resequencing protocol provides a scalable sample-handling tool for a genetic variation study of lung adenocarcinoma.
Acknowledgments
This work was partly supported by the Industrial Strategic Technology Development Program, 10040231, "Bioinformatics platform development for next-generation bioinformation analysis," funded by the Ministry of Knowledge Economy (MKE, Republic of Korea). We thank Drs. Kim and Lee of Samsung Medical Center (Seoul, Republic of Korea) for providing the cancer cells.
References
Supplementary materials
Supplementary data including two tables can be found with this article online at http://www.genominfo.org/src/sm/gni-12-50-s001.pdf.
Supplementary Table 1
Single nucleotide variations (SNVs) comparison between normal and cancer samples
Supplementary Table 2
Short insertions and deletions (INDELs) comparison between normal and cancer samples