Introduction
Next-generation sequencing (NGS) has resulted in an exponential growth in data in the past decade, primarily fueled by the general interest of the scientific community in functionally annotating the human genome. The power of high-throughput parallel sequencing was deployed to characterize human genetic variation, in an international effort launched in 2008, called the 1000 Genomes Project, with the hope that such a large-scale endeavor would help in identifying all the underlying genetic differences that lead to disease resistance/susceptibility [1].
In the meantime, based on numerous genome-wide association studies (GWAS), it was realized that 93% of the trait-associated polymorphisms within the human population were located in non-coding regions, potentially within cis-regulatory elements [2]. In fact, in the mid-1970s, King and Wilson [3] pointed out the importance of mapping regulatory events and hinted that a small difference in the time/level of activation of a single gene could influence overall systemic development. They estimated the genetic distance between human and chimpanzee to be much smaller than the corresponding genetic distance between the sibling species of Drosophila, thereby underscoring the need to explore regulatory/expression differences at different times as a basis to explain the evolutionary differences at the organismal level [3].
Why the Epigenome?
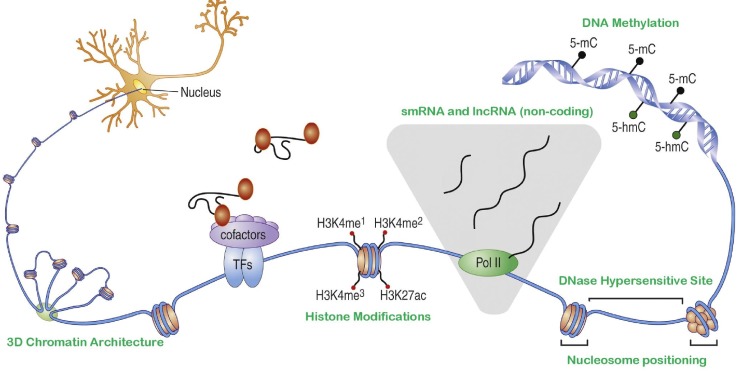
A mechanistic understanding of spatio-temporal gene expression in eukaryotes is far from complete. While genetic differences might be expected to explain expression divergence across species and expression variability across individuals of a population, it cannot explain how almost-identical genomes in more than 200 different cell types within an individual organism can drive such varied expression profiles specific to each cell type [4]. DNA is packaged in a nucleoprotein complex structure called chromatin that is highly dynamic, in that its "states" vary from one cell type to another. Therefore, a possible solution to this conundrum is to analyze these "states" at the genome level, i.e., characterize the landscape of the epigenome, which further qualifies and modulates the functional effect of genetic information [5]. Epigenome, meaning "on or above genomes," refers to such sequence-independent heritable properties of the genome that can modulate the functional output of the genome. While, as described initially, heritability is an important qualifying criterion of an epigenomic mark (most notably, DNA methylation) [6], currently, the term epigenome is loosely used to encompass a myriad of chemical changes to DNA or histone proteins, chromatin accessibility, small and long (non-coding) RNA localization, and higher-order DNA organization (including nucleosome occupancy and positioning, and 3D chromatin interactions) (Fig. 1) [7,8,9]. The ultimate combined effect of the epigenome is to determine the transcriptome (the set of all transcripts) of a cell precisely. Epigenomic features influence the regulatory program of each gene's expression in several ways: they define the local environment of specific processes by regulating the chromatin architecture, determine access of transcription factors to DNA, as well as serve to keep a "memory" of cell type-specific features facilitating heritability of epigenetic characteristics [10, 11].
The Wild West of Epigenetic Marks and the Impact of NGS Techonologies
The will of the global scientific community to sequence the human genome spurred advances in sequencing technologies that led to what is now known as NGS. The improvement in cost and speed of sequencing in the last decade has significantly surpassed the analogous improvement in computer technologies as predicted by Moore's law [12]. Well beyond the initial goal of sequencing complete genomes of several species, NGS is now routinely used as a tool to investigate the diverse array of cellular events involving DNA or RNA, such as the identification of genomic loci bound by a specific protein, the detection of pairs of spatially proximal or interacting chromosomal loci, etc. Broad availability of NGS technologies has led to a paradigm shift in molecular biology research; from probing singular cellular events to an unbiased genome-scale mapping of such events, and has enabled genome-wide elucidation of the epigenomic landscapes in hundreds of cell types, across developmental times, in human, as well as other species [13].
As mentioned earlier, it is the locus-specific "epigenetic code" (a specific signature or combination of several epigenetic modifications) that helps define a cell's expression program and identity [14], thereby distinguishing it from other cell types. Unlike the largely static genome of an individual, the epigenome is highly variable and dynamic; yet, many of these marks can be passed down a cell's lineage, or from one generation to the next. In this section, we describe several well-studied epigenomic marks and outline what is known about their role in gene expression modulation. We will also outline various NGS-based assays that are used in comprehensively mapping each of the epigenetic marks (Table 1) [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34].
DNA methylation
One of the more stable and heritable epigenetic marks is DNA methylation. The human genome is highly methylated; approximately 80% of cytosines in CpG dinucleotides are chemically modified at their fifth carbon atom with a methyl group [35]. Historically, DNA methylation was associated with transcriptional silencing (as evidenced by many promoter-based studies) [36]. Although genome-scale profiling of this epigenetic mark revealed that in some instances DNA methylation is correlated with transcriptional activation, it is enriched in the gene bodies of active genes [37]. Thus, the associations between an epigenomic mark and functional output may be location-specific, thereby complicating their functional interpretation.
The available assays for identifying methylated CpG dinucleotides in a genome vary in terms of resolution and cost. Restriction enzyme-based assays (e.g., methylation-sensitive restriction enzyme sequencing) involve the digestion of genomic DNA with several methylation-sensitive enzymes [15], followed by sequencing of digested fragments. Affinity-based enrichment assays, such as methylated DNA immunoprecipitation sequencing and methyl-CpG-binding domain protein sequencing [16, 17], selectively target methylated fragments (usually up to 100 bp) using an antibody, followed by sequencing of those fragments. Both these methods are low-resolution and essentially qualitative. Chemical modification methods, such as bisulfite sequencing (BS), have higher resolution and provide quantitative estimates of methylation. They exploit the fact that when treated with sodium bisulfite, unmethylated cytosines convert to uracil while methylated cytosines do not. Thus, mapping of sequenced fragments reveals the quantitative estimate of methylation at each CpG locus. The two variations to BS include reduced representation BS and whole-genome bisulfite sequencing, or methylC-seq (whole-genome BS); the former employs restriction enzyme digestion to target certain regions of interest prior to sodium bisulfite treatment [18], whereas the latter couples the chemical treatment with whole-genome sequencing [19].
Upon the discovery that 5-hydroxymethylcytosine (5-hmC: an intermediate produced during active DNA demethylation) and 5-methylcytosine (5-mC) are both resistant to bisulfite treatment, it became critical to distinguish between the different methyl variants, including 5-formylcytosine and 5-carboxylcytosine. To this end, oxidative bisulfite sequencing (oxBS-seq) was invented as an improvement for methylC-seq to measure single base-pair resolution methylation levels of 5-mC [20]. This is accomplished by oxidizing 5-hmCs (using potassium perruthenate) before sodium bisulfite treatment, such that they are now sensitive to the subjected chemical conversion to uracil, thus ensuring that the remaining cytosines are all 5-mCs.
Histone modifications
Chromosomal DNA is wrapped around histone octamers, essentially composed of 4 kinds of subunits; viz., H2A, H2B, H3, and H4. These proteins are subject to chemical modifications at specific residues of histone tails; some well-studied modifications include phosphorylation, methylation, and ubiquitination [38]. These modifications are involved in setting the stage for directed transcriptional activation and repression by controlling DNA accessibility or recruitment of other protein complexes. This idea has been extended into the "histone code" hypothesis [39, 40]-that complex combinations of distinct histone modifications, like H3K27me3 (a mark of repressed regions), H3K4me3 (a mark of gene promoters), and H3K27ac (a mark of transcriptionally active regions), etc. [41], underlie specific transcriptional programs. This notion has been further extended in more recent works into an 'epigenomic code' to include epigenomic marks other than histone modifications [42].
Chromatin immunoprecipitation (ChIP) of DNA-histone complexes is achieved by cross-linking histones with the DNA, digesting the cross-linked DNA, and then using an antibody specific to the N-tail modification of interest (to any of the >100 post-translational modifications to histone tails and globules [43]), followed by sequencing of the purified DNA fragments. This method is called chromatin immunoprecipitation sequencing (ChIP-seq) and was primarily used to map the genomic locations of DNA-associated proteins. Due to the relatively large fragment size, the mapped sequence reads thus obtained from ChIP-seq [21] provide a low-resolution localization map of modified histones in the genome. An alternative strategy, chromatin immunoprecipitation-exonuclease (ChIP-exo), uses exonuclease to digest the precipitated DNA fragment from either end to better resolve the modified loci at base-pair resolution [22].
Nucleosome positioning and occupancy
The packaging of DNA coiled around histones produces distinct structures called nucleosomes that form repeating units with approximately 147 bp wrapped around each unit, separated by varying lengths of linker DNA. The occupancy and periodic positioning of nucleosomes can control the accessibility of DNA to transcription factors [23] and DNases, as well as the transcription rate of active gene bodies [44], and are thus considered an epigenetic mark. Biochemically active regulatory regions are generally depleted of nucleosomes [45], whereas inactive repeat regions (heterochromatin) have higher affinity to form nucleosome structures [46].
A map of nucleosome occupancy is generated by using micrococcal nuclease (MNase) for digestion of the chromatin [23], followed by high-throughput sequencing-a technique called MNase-seq. MNase digests all linker DNA but preserves the DNA wound around the nucleosome; when the latter is sequenced and mapped to the reference, a map of the original positioning of nucleosomes is obtained. Due to some inherent biases in MNase affinity (preference to AT-rich regions) and the lack of single base-pair resolution data, attempts were made to develop more precise technologies [24]. An MNase-independent technique that involves chemically modifying engineered histones to bring about cleavage of DNA wound around histones allows direct mapping of nucleosome centers.
Chromatin accessibility
Epigenetic mechanisms, such as chromatin accessibility, impact transcription factor binding to DNA, transcriptional specificity, and hence, transcriptional regulation. Open and easily accessible regions of DNA within the chromatin are indicative of local territories of transcriptional activity. Measuring "openness" of DNA at different regions genome-wide, based on the DNase hypersensitivity assay, has helped discover several classes of functional elements, like promoters and enhancers [47]. It has also aided in identifying cell-type specific behaviors by comparison of accessibility profiles [47].
Regions of accessible chromatin are reflective of active regulatory sites. The enzyme DNase I is capable of digesting DNA in nucleosome-depleted regions (i.e., free unwound DNA). Post-digestion sequencing reveals large blocks of DNase hypersensitive sites (DHS) in chromatin (DNase-seq) [25, 48], which, upon further deep sequencing, can reveal up to 40-bp footprints of protected regions (potentially bound by transcription factors). These smaller regions are called digital genomic footprints (DGF) [26]. Formaldehyde-assisted isolation of regulatory elements sequencing is another technique that, by deep-sequencing random fragments of the genome post-crosslinking, measures the frequency of shearing at different loci, thus quantifying accessibility at high resolution, because free DNA that is unbound by factors is more susceptible to shearing [27].
3D chromatin architecture
The array of nucleosomes organized in 30-nm fibers, called chromatin, has diverse functions well beyond mere compaction. Coordinated activity of distal elements is orchestrated by short- and long-range DNA interactions, which is determined by the 3D chromatin structure as well as the local environment of individual genes. For instance, chromatin conformation/looping mediates a promoter's access to its enhancers, thereby determining the transcriptional fate of a gene [49]. Spatial proximity can be determined by measuring the interaction frequencies of linearly distal fragments by a suite of chromosome conformation capture (3C)-based assays.
Chromosomes assume a specific tertiary structure with profound implications for cellular function and fate [49]. While the overall chromatin structure cannot be directly measured, measuring spatial distances between pairs of genomic loci has become increasingly more efficient with the arrival of 3C technologies [28]. Treatment with formaldehyde crosslinks spatially proximal DNA loci in cells, and then post-digestion, the resulting fragments are allowed to ligate in a weak solution (cross-linked fragments ligate with higher frequency than random fragments). This frequency is quantitatively assessed by quantitative polymerase chain reaction. Known loci are selected for by using sequence-specific probes that allow assaying specific pairs of regions in so-called one-versus-one mapping. Circular chromosome conformation capture, or 4C, allows whole-genome mapping of all spatial interactions of one specific region of interest (one-vs-all mapping) [29]. Chromosome conformation capture carbon copy, or 5C, deploys several anchors and primers to ensure targeting of several regions at once to attain higher coverage (many-vs-many) [30]. Finally, the most recent derivative of 3C, namely Hi-C, permits surveying of the whole genome [31] in a relatively unbiased fashion for frequencies of spatially interacting pairs of loci in the entire genome without restricting the exploration to selected loci. This was done with reasonably high resolution, where the human genome was partitioned into 100-kb blocks and the spatial interaction between every pair of intra-chromosomal regions was assessed, while interchromosomal interactions have been mapped at 1-Mb resolution [31].
A variation of Hi-C, called chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), involves a ChIP reaction to isolate chromatin interactions involving regions of DNA that bind to a protein of interest [32]. For example, an antibody targeting RNA polymerase II can be used to precipitate interactions involving a promoter and another interacting region, such as a distal enhancer or other promoters [50].
Non-coding RNA localization
Specific classes of non-coding RNA-short RNAs (viz. micro RNA [miRNA], short interfering RNA [siRNA], and piwi-interacting RNA [piRNA]) and long non-coding RNAs (lncRNA)-regulate gene expression through epigenetic mechanisms, influencing several cellular processes, like X chromosome inactivation [51], genomic imprinting [52], and some cellular processes [53], and cancer [54]. LncRNAs tether epigenetic complexes capable of methylating DNA and modifying histones to the chromatin, enabling allele- and locus-specific regulation. For example, lncRNA Xist covers the inactive X chromosome and recruits PCR2, a Polycomb complex responsible for trimethylation of H3K27 [51]. Furthermore, lncRNAs, by virtue of their length, are suited for orchestrating specific regulatory events particular to a target locus [55].
Additionally, miRNA (~22 nucleotides long) have shown to be linked with cancer and can act as either oncogenes or tumor-suppressor genes. They can be involved in establishing DNA methylation and regulating histone modification by interacting with their target mRNA to alter chromatin of the corresponding DNA template [56]. siRNA, and its interference machinery has also been implicated in the formation of heterochromatin [57].
A catalog of small RNAs has been generated by deep sequencing of total RNA from whole transcriptomes using smRNA-seq technology [34]. Selection of smRNA can be carried out by sequential ligation of adapters to its unique 5' mono/triphosphate ends, produced as a result of processing the smRNA population in the cell [58]. On the other hand, lncRNA are poly-adenylated, and much of the challenge in identifying them from whole-genome mRNA-seq data [33] lies in the computational and analytical domains [59].
Epigenome Projects and Consortia
Spurred by unprecedented advances in sequencing technologies as well as other assays, and a recognition of the potential of NGS in determining the epigenomic landscape, several research consortia have formed to generate genome-wide maps of human epigenomic marks by sharing resources and publishing standard operating procedures, thereby ensuring best practices and high-quality datasets [60]. One of the first concerted efforts in this direction was the Encyclopedia of DNA Elements (ENCODE) Project, started in 2003 and funded by the National Human Genome Research Institute at the National Institute of Health (NIH-NHGRI), which involved hundreds of researchers in dozens of labs across the globe. The ENCODE consortium has performed 1,650 epigenomic profiling on 147 cell lines assessing the transcriptome, transcription factor binding for dozens of transcription factors, chromatin topology, histone modifications, DNA methylation, and more (http://www.encodeproject.org) [13]. Five years after launching ENCODE, the NIH funded a second large-scale mapping project called the Roadmap Epigenomics Program. While ENCODE focused on generating data in cell lines, the Roadmap program focuses on mapping epigenomes of high-priority normal human primary tissue and human embryonic stem cell (ESC)-derived cell types. As of May 2012, the Roadmap includes 61 "complete" epigenomes (of a variety of cell types) (http://www.roadmapepigenomics.org) [61]. The data repository can be accessed interactively at Human Epigenome Atlas (http://www.genboree.org/epigenomeatlas/multiGridViewerPublic.rhtml), via the Washington University Epigenome Browser (http://www.epigenomegateway.wustl.edu/browser), as well as the UCSC Genome Browser (http://www.genome.ucsc.edu). Further, the NIH Roadmap is one of the members of an even larger consortium called the International Human Epigenome Consortium (IHEC; http://www.ihec-epigenomes.org), formed by institutions in 7 countries. To name a few, BLUEPRINT: a European initiative to generate about 100 reference epigenomic maps; the CREST/IHEC team in Japan: to produce reference epigenomes specializing in gastrointestinal epithelial cells, vascular endothelial cells, and cells of reproductive organs; and the Epigenomic Platform Program: a Canadian program to map epigenomic variations in disease cells. Together, these efforts are expected to generate over 1,000 reference human epigenomes. Other epigenome mapping efforts focused on specific disease areas have also been undertaken. These include the Cancer Genome Atlas (TCGA), funded by the NIH in 2006 [62], which is a part of the International Cancer Genome Consortium (ICGC; http://www.icgc.org) that was officially put together in 2008.
What Have We Learned?
By providing a rich functional annotation of the genome, the numerous epigenomic datasets have significantly improved our ability to probe the mechanisms of gene regulation as well as shed light on disease processes mediated by perturbation of normal regulatory processes. A collection of 30 scientific reports published with the initial release of the ENCODE data provides a detailed report of the initial integrative analysis of the epigenomics data [47, 63, 64, 65, 66].
At a basic level, the data have revealed epigenomic signatures of a variety of functional elements, thus significantly enriching their annotation. For instance, many non-coding RNAs have been successfully annotated as a result of deciphering an epigenomic code specific to their promoters and gene bodies [67, 68]. High-throughput profiling of the histone modification HeK4me1 has revealed that it is a characteristic mark of enhancer elements; this, and other correlative marks have enabled the discovery of tens of thousands of cell-specific enhancers [69, 70] in the human genome, drastically improving our ability to probe the mechanisms of transcriptional regulation. In fact, chromatin signatures specific to promoters, enhancers, and repressed regions, etc., have all been modeled by computational methods like ChromHMM [42]. Furthermore, by isolating footprints of protected DNA present inside blocks of hypersensitive or nucleosome-depleted DNA, a comprehensive list of potential cis-regulatory binding sites/sequences has been identified, despite the lack of specific antibodies or the knowledge of cell-specific regulators [2].
Availability of epigenomic data has revived predictive modeling of condition-specific gene expression. For example, Dong et al. [71] generated a 2-step quantitative model that includes genome localization data for 11 histone modifications and 1 histone variant in 7 human cell lines and gene expression data quantified using RNA-seq and cap analysis gene expression (CAGE) in the nuclear and cytosolic compartments of the cell. The Pearson's correlation coefficient between their predicted and measured expression levels in about 78 experiments ranged between 0.6 and 0.9 (median r = 0.83). Natarajan et al. [72] trained a classifier that associates cell-specific TF sequence motif matches in DHS regions with different expression patterns in 19 diverse cell types, thus predicting cell-type specific expression directly from regulatory elements in open chromatin.
Prior to the availability of NGS, details of the spatial structure of chromatin and its correlates, as well as its functional impacts were largely uncharted. Characterization of the spatial proximity of distal genomic elements with 3C-derivative assays has revealed a high rate of widespread promiscuous interactions among enhancers and promoters. For instance, more than 40% of promoters interact with multiple distal sites, and this is likely true for enhancers as well [73]. The spatial chromatin interaction data have also helped challenge some previously held beliefs that were nevertheless unsupported. For instance, historically, the DNA-binding protein CTCF was known to function, among other things, as an insulator that inhibited the spatial interaction between an enhancer and its target promoter when flanking a CTCF-bound locus. However, recent 5C data showed that almost 60% of all enhancer-promoter interactions happen, despite an intervening CTCF-bound locus [74]. In fact, there is increasing evidence that CTCF, along with cohesin is involved in coordinating long-range interactions between regulatory elements, and that these events are reflective of cell-type specific transcriptional programs [75].
With the availability of putative genome-wide enhancers along with their cell type-specific activity profile across numerous cell types, it is now possible to investigate an alternate layer of transcriptional regulation represented by a network of distal enhancers that exhibit correlated activities across cell types [76, 77] and jointly underlie correlated expression of functionally linked genes. Such analysis brings forth an alternative view of transcriptional regulation, where instead of a single gene regulated by one or more regulatory elements, one ought to consider the collective of enhancers and genes, co-localized in nuclear space to achieve coexpression of functionally linked genes [78, 79].
Epigenomic profiling along the developmental time course from ESCs to differentiated cells has shed light on transcriptional regulatory mechanisms during early development. For instance, comparison of the "histone code" between the two developmental timeframes indicates that regions marked by repressive histone marks are generally larger in differentiated cells and that these regions span developmental genes. It was also noted that the same regions are more accessible in ESCs [80, 81], thus suggesting an epigenome-mediated regulation of developmental gene transcription. Precisely how this is accomplished is yet to be clarified.
The epigenomic data and the discovery of significant patterns of epigenomic marks associated with specific functional regions have also helped clarify genotype-phenotype association data. Numerous GWAS have revealed several polymorphic loci associated with various diseases [82]. However, a mechanistic interpretation of these signals has been hampered by the fact that most of the disease-associated genetic variants reside in non-coding regions with no obvious functional interpretation. By combining epigenomic maps and genetic variants, it was found that these disease-associated variants are enriched within cell type-specific accessible (DHS regions) and active regulatory regions [2]. Also, an epigenomic model of cardiac enhancers was shown to provide a better causal interpretation of GWAS signals associated with heart-related traits [83].
Open Questions and Challenges
The slew of epigenetic data has revolutionized our perception of the human genome. Although not all "epigenetically marked" regions of the genome are likely to be functional, it is very likely that a large portion is functional-much more than the 5-10% of the genome that is deemed to evolve under purifying selection [84, 85]. Analysis of transcriptional and epigenetic data has revealed that almost half of the genome is involved in carrying out specific biochemical functions [13]. Where these biochemical functions fit in the big picture, and how they are regulated are still open questions.
Another fundamental problem is that the causal relationships among various epigenomic events, such as DNA methylation, histone modifications, TF binding, and gene expression, etc., are currently not well understood. Although correlations between these events have been well documented in different contexts, we do not know if one is necessary and/or sufficient to observe another.
Before the full potential of NGS in mapping the epigenome can be attained, several technical challenges need to be overcome. For instance, techniques like ChIP-seq, used for profiling transcription factor binding and histone modifications require a large amount of starting material, ranging up to 5 million cells. Collection of such a large population of primary differentiated cells, progenitors, and those from specific developmental stages is inherently hard. Heterogeneity of a cell population is another issue that needs to be resolved. It is challenging to obtain a homogeneous population of cells of a singular type from primary tissues that often contain a heterogeneous population of cell types, and it is this lack of homogeneity that can adversely impact the interpretability and generalizability of the results obtained from analyzing the epigenomic data.
Yet, many solutions to tackle the problems above are underway. First, there is the emergence of techniques-like ChIP-nano (nano-scale ChIP), which is a highly sensitive small-scale ChIP-seq protocol with a requirement of <50,000 cells as starting material [86], and single-molecule real-time sequencing [87], which aims at alleviating cell-population effects during mapping specific features. Second, methods that are aimed at the targeted knock-out of epigenetic modifiers, like DNA methyltransferases and histone modification enzymes, at predetermined DNA sequences (using transcription activator-like effector nucleases [TALENs]) [88], can help to better characterize the functional relationships between gene expression and epigenetics, thus providing proof of concept for expression modulation (also called epigenetic editing methods) [89]. Also, powerful statistical approaches are being developed to integrate data [42, 90] to resolve some of these fundamental issues. With more epigenomic maps being profiled, and more effective computational approaches being developed, we are inching towards a holistic mechanistic understanding of cellular and organismal processes.