Introduction
Human leukocyte antigen (HLA) in the major histocompatibility complex (MHC) region is an important genetic factor for a wide range of human diseases, such as type 1 diabetes [1] and rheumatoid arthritis [2, 3]. In association studies of HLA, it is essential to investigate the high-resolution genotypes of HLA genes. However, genotyping HLA alleles in a large number of samples can be prohibitively expensive due to the high HLA typing cost. To overcome this challenge, imputation approaches have been recently developed to impute HLA alleles from intergenic single nucleotide polymorphism (SNP) data [4-6]. These approaches, of which SNP2HLA [4] is widely used, can impute high-resolution HLA alleles efficiently and have been utilized in a number of large-scale fine-mapping HLA analyses of diseases [1-3, 7, 8].
Although the use of imputation approaches has become prevalent, imputation errors can always be present. To reduce imputation errors, it is often necessary to use a large reference panel that can capture diverse HLA alleles. Both the size of the panel and the diversity of alleles can increase if we can use multiple reference panels at the same time. However, in current HLA imputation frameworks, there is no streamlined pipeline for simultaneously utilizing multiple reference panels.
In this study, we present a software package, MergeReference, that merges multiple reference panels in SNP2HLA [4] format into a single panel for HLA imputation. By merging multiple reference panels of similar ethnicities, we can increase both the sample size and the allele diversity of the panel and therefore improve the imputation accuracy. We compared the performance of the Pan-Asian reference panel [9], the Korean reference panel [10], and the merged reference panel generated by our software by masking and imputing the HLA alleles in the HapMap [11] Asian dataset. The merged panel achieved an average 4-digit accuracy of six HLA genes of 93.9%, whereas the Pan-Asian and the Korean panels had 86.6% and 91.5% accuracy rates, respectively, demonstrating that the simultaneous use of multiple reference panels improves the accuracy. MergeReference is freely available at http://software.buhmhan.com/MergeReference.
Methods
MergeReference
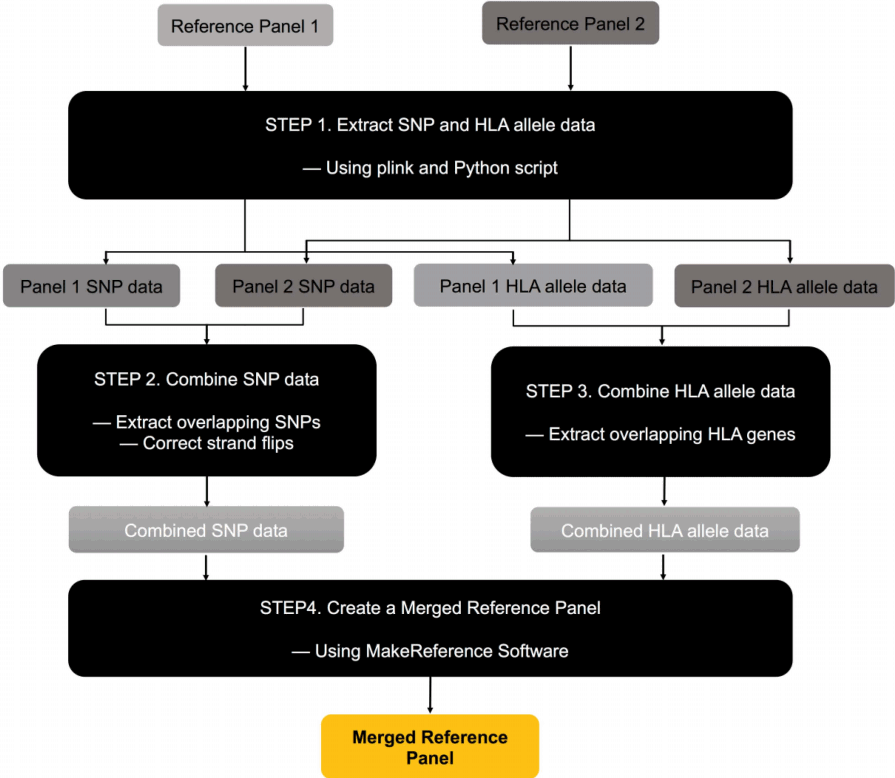
MergeReference is a software package that merges multiple reference panels for HLA imputation. We assume that the panels are in SNP2HLA format [4]. MergeReference consists of the following four steps (Fig. 1). Although these steps are for merging two panels, merging more than two panels can be done straightforwardly by repeatedly applying the same procedure to pairs of panels until all panels are merged into one.
Step 1. Extracting SNP and HLA allele data
Given two reference panels to be merged, we extract SNP data from them using PLINK [12]. Then, we extract HLA genotype information from the reference panel. Because each allele in an HLA gene is coded as a binary marker in SNP2HLA format, extracting HLA allele information requires the examination of the entire set of binary markers for an HLA gene. We use our Python script to perform this extraction.
Step 2. Combining SNP data
In this step, we combine the SNP data of the two panels. First, we select overlapping SNPs that exist in both panels. An alternative approach would be to keep all SNPs as partially missing data. However, in this alternative approach, the missing values in this step will be automatically imputed in step 4 and will be used as reference data in the imputation. Because using imputed data as reference data can degrade the performance, we avoided this alternative approach. Second, we check and correct the strand flips of the SNPs between the two panels. For A/T and G/C SNPs with strand flips that are difficult to detect, we use frequency information.
Step 3. Combining HLA data
We combine the HLA data of the two panels. As we did in step 2, in order to avoid using imputed values as reference data, we only keep overlapping HLA genes that exist in both panels.
Step 4. Creating a merged reference panel
Given the combined SNP data from step 2 and the combined HLA data from step 3, we employ MakeReference software in the SNP2HLA package [4] to construct the new merged reference panel in SNP2HLA format.
Results
Merging Korean and Pan-Asian panels
To test our MergeReference software, we merged the Pan-Asian panel (n=441) [9] and the Korean panel (n=413) [10]. The Korean panel included 4526 SNPs, and the Pan-Asian panel included 4,603 SNPs in the MHC region (chr6:25-34Mb). The Korean panel had genotype information on six HLA genes (A, B, C, DRB1, DPB1, and DQB1), and the Pan-Asian panel had genotype information on eight HLA genes (A, B, C, DRB1, DPA1, DPB1, DQA1, and DQB1). After applying MergeReference, 2,995 overlapping SNPs and the genotype information of six overlapping HLA genes were left. Merging the two panels took less than 10 min on a standard computer (2.7 GHz Intel Core i5 CPU, 8 GB memory).
Merged panel can improve performance
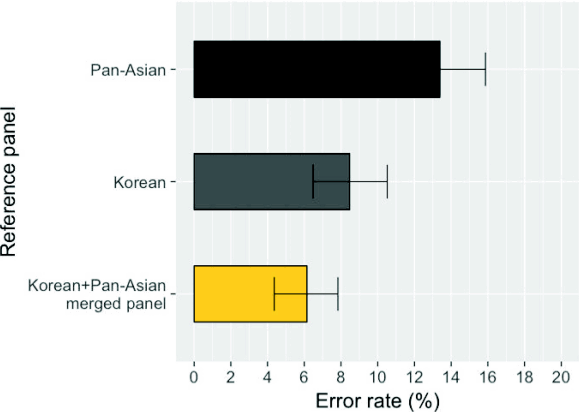
To compare the imputation accuracy of different panels, we used the 61 HapMap Asian samples from Beijing, China (CHB) and Tokyo, Japan (JPT) as benchmark data [11]. We masked the HLA information of these individuals, imputed them, and compared the imputed genotypes to the true genotypes. We evaluated three panels: the Pan-Asian panel, the Korean panel, and the merged panel of the two. The merged panel achieved an average 4-digit accuracy of six HLA genes of 93.8%, whereas the Pan-Asian and Korean panels had 85.4% and 91.5% accuracy rates, respectively (Fig. 2). The per-HLA gene accuracies are described in Table 1. The performance of the merged panel was equal or superior to that of the individual panels for all HLA genes. The improvement in performance was notable for HLA-DRB1 (Pan-Asian, 85.2%; Korean, 89.3%; merged, 95.1%).
Discussion
We have developed MergeReference, a software package that provides a streamlined pipeline for combining multiple reference panels into one for HLA imputation. In a previous study, Kim et al. [13] also evaluated the gain in performance of merging reference panels for HLA imputation, although they did not provide a software pipeline. Similar to our work, their results supported the utility of merging panels, particularly when the ethnicities are similar. However, merging Asian panels with a European panel did not provide better performance for imputing HLA in Asian samples [13]. Therefore, determining whether merging multiple panels increases the accuracy depends on how similar the ethnicities of the panels are. If it is unclear whether merging panels would help or not, it can be good practice to evaluate the approximate accuracy by using a public dataset, such as HapMap, or setting aside a subset of the panel samples as a test dataset. Another issue that can affect the performance of the merged panel is the sample size balance. We found that if one panel is much larger (e.g., by 10-fold) than the other panel, the merged panel often has essentially the same performance as the single large panel. In such situations, subsampling from the large panel may improve the balance of the two panels, despite the decreased sample size. Further investigations will be needed to thoroughly explore these issues related to the gain in performance with merging, and we expect that our software pipeline will facilitate such explorations in future studies.