References
2. Centers for Disease Control and Prevention. Hearing loss in children Atlanta: Center for Disease Control and Prevention; 2018. Accessed 2018 Nov 20. Available from:
https://www.cdc.gov
.
3. Bruns L, Murbe D, Hahne A. Understanding music with cochlear implants. Sci Rep 2016;6:32026.
4. Huang J, Sheffield B, Lin P, Zeng FG. Electro-tactile stimulation enhances cochlear implant speech recognition in noise. Sci Rep 2017;7:2196.
5. Morton CC, Nance WE. Newborn hearing screening: a silent revolution. N Engl J Med 2006;354:2151–2164.
6. Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage The Authors: Hereditary Hearing Loss Homepage; 2018. Accessed 2018 Nov 20. Available from:
https://www.hereditaryhearingloss.org
.
7. Guilford P, Ben Arab S, Blanchard S, Levilliers J, Weissenbach J, Belkahia A, et al. A non-syndrome form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat Genet 1994;6:24–28.
8. Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80–83.
9. del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002;346:243–249.
10. Guilford P, Ayadi H, Blanchard S, Chaib H, Le Paslier D, Weissenbach J, et al. A human gene responsible for neurosensory, non-syndromic recessive deafness is a candidate homologue of the mouse sh-1 gene. Hum Mol Genet 1994;3:989–993.
11. Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, et al. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet 1997;16:188–190.
12. Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, et al. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 1997;16:191–193.
13. Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, et al. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet 1995;9:86–91.
14. Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3
. Science 1998;280:1447–1451.
15. Baldwin CT, Weiss S, Farrer LA, De Stefano AL, Adair R, Franklyn B, et al. Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum Mol Genet 1995;4:1637–1642.
16. Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, et al. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet 1998;18:215–217.
17. Fukushima K, Ramesh A, Srisailapathy CR, Ni L, Chen A, O’Neill M, et al. Consanguineous nuclear families used to identify a new locus for recessive non-syndromic hearing loss on 14q. Hum Mol Genet 1995;4:1643–1648.
18. Fukushima K, Ramesh A, Srisailapathy CR, Ni L, Wayne S, O’Neill ME, et al. An autosomal recessive nonsyndromic form of sensorineural hearing loss maps to 3p-DFNB6. Genome Res 1995;5:305–308.
19. Naz S, Giguere CM, Kohrman DC, Mitchem KL, Riazuddin S, Morell RJ, et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet 2002;71:632–636.
20. Jain PK, Fukushima K, Deshmukh D, Ramesh A, Thomas E, Lalwani AK, et al. A human recessive neurosensory nonsyndromic hearing impairment locus is potential homologue of murine deafness (dn) locus. Hum Mol Genet 1995;4:2391–2394.
21. Scott DA, Carmi R, Elbedour K, Yosefsberg S, Stone EM, Sheffield VC. An autosomal recessive nonsyndromic-hearing-loss locus identified by DNA pooling using two inbred Bedouin kindreds. Am J Hum Genet 1996;59:385–391.
22. Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet 2002;30:277–284.
23. Veske A, Oehlmann R, Younus F, Mohyuddin A, Muller-Myhsok B, Mehdi SQ, et al. Autosomal recessive non-syndromic deafness locus (DFNB8) maps on chromosome 21q22 in a large consanguineous kindred from Pakistan. Hum Mol Genet 1996;5:165–168.
24. Bonne-Tamir B, DeStefano AL, Briggs CE, Adair R, Franklyn B, Weiss S, et al. Linkage of congenital recessive deafness (gene DFNB10) to chromosome 21q22.3. Am J Hum Genet 1996;58:1254–1259.
25. Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 2001;27:59–63.
26. Chaib H, Place C, Salem N, Chardenoux S, Vincent C, Weissenbach J, et al. A gene responsible for a sensorineural nonsyndromic recessive deafness maps to chromosome 2p22-23. Hum Mol Genet 1996;5:155–158.
27. Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, et al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 1999;21:363–369.
28. Chaib H, Place C, Salem N, Dode C, Chardenoux S, Weissenbach J, et al. Mapping of DFNB12, a gene for a non-syndromal autosomal recessive deafness, to chromosome 10q21-22. Hum Mol Genet 1996;5:1061–1064.
29. Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 2001;68:26–37.
30. Mustapha M, Chardenoux S, Nieder A, Salem N, Weissenbach J, el-Zir E, et al. A sensorineural progressive autosomal recessive form of isolated deafness, DFNB13, maps to chromosome 7q34-q36. Eur J Hum Genet 1998;6:245–250.
31. Mustapha M, Salem N, Weil D, el-Zir E, Loiselet J, Petit C. Identification of a locus on chromosome 7q31, DFNB14, responsible for prelingual sensorineural non-syndromic deafness. Eur J Hum Genet 1998;6:548–551.
32. Chen A, Wayne S, Bell A, Ramesh A, Srisailapathy CR, Scott DA, et al. New gene for autosomal recessive non-syndromic hearing loss maps to either chromosome 3q or 19p. Am J Med Genet 1997;71:467–471.
33. Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, et al. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Commun 2011;2:201.
34. Rehman AU, Gul K, Morell RJ, Lee K, Ahmed ZM, Riazuddin S, et al. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum Genet 2011;130:759–765.
35. Verpy E, Masmoudi S, Zwaenepoel I, Leibovici M, Hutchin TP, Del Castillo I, et al. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat Genet 2001;29:345–349.
36. Greinwald JH Jr, Wayne S, Chen AH, Scott DA, Zbar RI, Kraft ML, et al. Localization of a novel gene for nonsyndromic hearing loss (DFNB17) to chromosome region 7q31. Am J Med Genet 1998;78:107–113.
37. Jain PK, Lalwani AK, Li XC, Singleton TL, Smith TN, Chen A, et al. A gene for recessive nonsyndromic sensorineural deafness (DFNB18) maps to the chromosomal region 11p14-p15.1 containing the Usher syndrome type 1C gene. Genomics 1998;50:290–292.
38. Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, et al. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum Genet 2002;111:26–30.
39. Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, et al. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet 2002;110:527–531.
40. Schraders M, Ruiz-Palmero L, Kalay E, Oostrik J, del Castillo FJ, Sezgin O, et al. Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am J Hum Genet 2012;91:883–889.
41. Green GE, Wayne S, Nishtala R, Chen AH, Ramesh A, Srisailapathy CR, et al. Identification of a novel locus (DFNB19) for non-syndromic autosomal-recessive hearing loss in a consanguineous family. In : Molecular Biology of Hearing and Deafness Meeting; 1998 Oct 8; Bathesda, MD.
42. Moynihan L, Houseman M, Newton V, Mueller R, Lench N. DFNB20: a novel locus for autosomal recessive, non-syndromal sensorineural hearing loss maps to chromosome 11q25-qter. Eur J Hum Genet 1999;7:243–246.
43. Mustapha M, Weil D, Chardenoux S, Elias S, El-Zir E, Beckmann JS, et al. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum Mol Genet 1999;8:409–412.
44. Zwaenepoel I, Mustapha M, Leibovici M, Verpy E, Goodyear R, Liu XZ, et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci U S A 2002;99:6240–6245.
45. Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 2003;12:3215–3223.
46. Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, et al. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 2007;28:417–423.
47. Schraders M, Lee K, Oostrik J, Huygen PL, Ali G, Hoefsloot LH, et al. Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am J Hum Genet 2010;86:138–147.
48. Yousaf R, Ahmed ZM, Giese APJ, Morell RJ, Lagziel A, Dabdoub A, Wilcox ER, Riazuddin S, Friedman TB, Riazuddin S. Modifier variant of METTL13 suppresses human GAB1-associated profound deafness. J Clin Invest 2018;128:1509–1522.
49. Pulleyn LJ, Jackson AP, Roberts E, Carridice A, Muxworthy C, Houseman M, et al. A new locus for autosomal recessive non-syndromal sensorineural hearing impairment (DFNB27) on chromosome 2q23-q31. Eur J Hum Genet 2000;8:991–993.
50. Shahin H, Walsh T, Sobe T, Abu Sa’ed J, Abu Rayan A, Lynch ED, et al. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet 2006;78:144–152.
51. Riazuddin S, Khan SN, Ahmed ZM, Ghosh M, Caution K, Nazli S, et al. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am J Hum Genet 2006;78:137–143.
52. Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 2001;104:165–172.
53. Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, et al. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A 2002;99:7518–7523.
54. Mustapha M, Chouery E, Chardenoux S, Naboulsi M, Paronnaud J, Lemainque A, et al. DFNB31, a recessive form of sensorineural hearing loss, maps to chromosome 9q32-34. Eur J Hum Genet 2002;10:210–212.
55. Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet 2003;34:421–428.
56. Masmoudi S, Tlili A, Majava M, Ghorbel AM, Chardenoux S, Lemainque A, et al. Mapping of a new autosomal recessive nonsyndromic hearing loss locus (DFNB32) to chromosome 1p13.3–22.1. Eur J Hum Genet 2003;11:185–188.
57. Delmaghani S, Aghaie A, Bouyacoub Y, El Hachmi H, Bonnet C, Riahi Z, et al. Mutations in CDC14A, encoding a protein phosphatase involved in hair cell ciliogenesis, cause autosomal-recessive severe to profound deafness. Am J Hum Genet 2016;98:1266–1270.
58. Medlej-Hashim M, Mustapha M, Chouery E, Weil D, Parronaud J, Salem N, et al. Non-syndromic recessive deafness in Jordan: mapping of a new locus to chromosome 9q34.3 and prevalence of DFNB1 mutations. Eur J Hum Genet 2002;10:391–394.
59. Ansar M, Din MA, Arshad M, Sohail M, Faiyaz-Ul-Haque M, Haque S, et al. A novel autosomal recessive non-syndromic deafness locus (DFNB35) maps to 14q24.1-14q24.3 in large consanguineous kindred from Pakistan. Eur J Hum Genet 2003;11:77–80.
60. Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, et al. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet 2008;82:125–138.
61. Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr, Khan SN, et al. Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet 2004;41:591–595.
62. Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet 2003;72:1315–1322.
63. Ansar M, Ramzan M, Pham TL, Yan K, Jamal SM, Haque S, et al. Localization of a novel autosomal recessive non-syndromic hearing impairment locus (DFNB38) to 6q26-q27 in a consanguineous kindred from Pakistan. Hum Hered 2003;55:71–74.
64. Schultz JM, Khan SN, Ahmed ZM, Riazuddin S, Waryah AM, Chhatre D, et al. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am J Hum Genet 2009;85:25–39.
65. Delmaghani S, Aghaie A, Compain-Nouaille S, Ataie A, Lemainque A, Zeinali S, et al. DFNB40, a recessive form of sensorineural hearing loss, maps to chromosome 22q11. 21-12.1. Eur J Hum Genet 2003;11:816–818.
66. Aslam M, Wajid M, Chahrour MH, Ansar M, Haque S, Pham TL, et al. A novel autosomal recessive nonsyndromic hearing impairment locus (DFNB42) maps to chromosome 3q13.31-q22.3. Am J Med Genet A 2005;133A:18–22.
67. Borck G, Ur Rehman A, Lee K, Pogoda HM, Kakar N, von Ameln S, et al. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am J Hum Genet 2011;88:127–137.
68. Ansar M, Chahrour MH, Amin Ud Din M, Arshad M, Haque S, Pham TL, et al. DFNB44, a novel autosomal recessive non-syndromic hearing impairment locus, maps to chromosome 7p14.1-q11.22. Hum Hered 2004;57:195–199.
69. Santos-Cortez RL, Lee K, Giese AP, Ansar M, Amin-Ud-Din M, Rehn K, et al. Adenylate cyclase 1 (ADCY1) mutations cause recessive hearing impairment in humans and defects in hair cell function and hearing in zebrafish. Hum Mol Genet 2014;23:3289–3298.
70. Bhatti A, Lee K, McDonald ML, Hassan MJ, Gutala R, Ansar M, et al. Mapping of a new autosomal recessive non-syndromic hearing impairment locus (DFNB45) to chromosome 1q43-q44. Clin Genet 2008;73:395–398.
71. Mir A, Ansar M, Chahrour MH, Pham TL, Wajid M, Haque S, et al. Mapping of a novel autosomal recessive nonsyndromic deafness locus (DFNB46) to chromosome 18p11.32-p11.31. Am J Med Genet A 2005;133A:23–26.
72. Hassan MJ, Santos RL, Rafiq MA, Chahrour MH, Pham TL, Wajid M, et al. A novel autosomal recessive non-syndromic hearing impairment locus (DFNB47) maps to chromosome 2p25.1-p24.3. Hum Genet 2006;118:605–610.
73. Ahmad J, Khan SN, Khan SY, Ramzan K, Riazuddin S, Ahmed ZM, et al. DFNB48, a new nonsyndromic recessive deafness locus, maps to chromosome 15q23-q25.1. Hum Genet 2005;116:407–412.
74. Ramzan K, Shaikh RS, Ahmad J, Khan SN, Riazuddin S, Ahmed ZM, et al. A new locus for nonsyndromic deafness DFNB49 maps to chromosome 5q12.3-q14.1. Hum Genet 2005;116:17–22.
75. Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, et al. Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet 2006;79:1040–1051.
76. Girotto G, Abdulhadi K, Buniello A, Vozzi D, Licastro D, d’Eustacchio A, et al. Linkage study and exome sequencing identify a BDP1 mutation associated with hereditary hearing loss. PLoS One 2013;8:e80323.
77. Shaikh RS, Ramzan K, Nazli S, Sattar S, Khan SN, Riazuddin S, et al. A new locus for nonsyndromic deafness DFNB51 maps to chromosome 11p13-p12. Am J Med Genet A 2005;138:392–395.
78. Chen W, Kahrizi K, Meyer NC, Riazalhosseini Y, Van Camp G, Najmabadi H, et al. Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J Med Genet 2005;42:e61.
79. Irshad S, Santos RL, Muhammad D, Lee K, McArthur N, Haque S, et al. Localization of a novel autosomal recessive non-syndromic hearing impairment locus DFNB55 to chromosome 4q12-q13.2. Clin Genet 2005;68:262–267.
80. Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet 2006;38:770–778.
81. Ben Said M, Grati M, Ishimoto T, Zou B, Chakchouk I, Ma Q, et al. A mutation in SLC22A4 encoding an organic cation transporter expressed in the cochlea strial endothelium causes human recessive non-syndromic hearing loss DFNB60. Hum Genet 2016;135:513–524.
82. Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, et al. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum Mol Genet 2003;12:1155–1162.
83. Ali G, Santos RL, John P, Wambangco MA, Lee K, Ahmad W, et al. The mapping of DFNB62, a new locus for autosomal recessive non-syndromic hearing impairment, to chromosome 12p13.2-p11.23. Clin Genet 2006;69:429–433.
84. Du X, Schwander M, Moresco EM, Viviani P, Haller C, Hildebrand MS, et al. A catechol-O-methyltransferase that is essential for auditory function in mice and humans. Proc Natl Acad Sci U S A 2008;105:14609–14614.
85. Ahmed ZM, Masmoudi S, Kalay E, Belyantseva IA, Mosrati MA, Collin RW, et al. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet 2008;40:1335–1340.
86. Tariq A, Santos RL, Khan MN, Lee K, Hassan MJ, Ahmad W, et al. Localization of a novel autosomal recessive nonsyndromic hearing impairment locus DFNB65 to chromosome 20q13.2-q13.32. J Mol Med (Berl) 2006;84:484–490.
87. Grati M, Chakchouk I, Ma Q, Bensaid M, Desmidt A, Turki N, et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet 2015;24:2482–2491.
88. Tlili A, Mannikko M, Charfedine I, Lahmar I, Benzina Z, Ben Amor M, et al. A novel autosomal recessive non-syndromic deafness locus, DFNB66, maps to chromosome 6p21.2-22.3 in a large Tunisian consanguineous family. Hum Hered 2005;60:123–128.
89. Shabbir MI, Ahmed ZM, Khan SY, Riazuddin S, Waryah AM, Khan SN, et al. Mutations of human TMHS cause recessively inherited non-syndromic hearing loss. J Med Genet 2006;43:634–640.
90. Kalay E, Li Y, Uzumcu A, Uyguner O, Collin RW, Caylan R, et al. Mutations in the lipoma HMGIC fusion partner-like 5 (LHFPL5) gene cause autosomal recessive nonsyndromic hearing loss. Hum Mutat 2006;27:633–639.
91. Santos RL, Hassan MJ, Sikandar S, Lee K, Ali G, Martin PE Jr, et al. DFNB68, a novel autosomal recessive non-syndromic hearing impairment locus at chromosomal region 19p13.2. Hum Genet 2006;120:85–92.
92. Santos-Cortez RL, Faridi R, Rehman AU, Lee K, Ansar M, Wang X, et al. Autosomal-recessive hearing impairment due to rare missense variants within S1PR2
. Am J Hum Genet 2016;98:331–338.
93. Chishti MS, Lee K, McDonald ML, Hassan MJ, Ansar M, Ahmad W, et al. Novel autosomal recessive non-syndromic hearing impairment locus (DFNB71) maps to chromosome 8p22-21.3. J Hum Genet 2009;54:141–144.
94. Riazuddin S, Anwar S, Fischer M, Ahmed ZM, Khan SY, Janssen AG, et al. Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am J Hum Genet 2009;85:273–280.
95. Waryah AM, Rehman A, Ahmed ZM, Bashir ZH, Khan SY, Zafar AU, et al. DFNB74, a novel autosomal recessive nonsyndromic hearing impairment locus on chromosome 12q14.2-q15. Clin Genet 2009;76:270–275.
96. Ahmed ZM, Yousaf R, Lee BC, Khan SN, Lee S, Lee K, et al. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet 2011;88:19–29.
97. Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, et al. The LINC complex is essential for hearing. J Clin Invest 2013;123:740–750.
98. Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, et al. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet 2009;85:328–337.
99. Rehman AU, Morell RJ, Belyantseva IA, Khan SY, Boger ET, Shahzad M, et al. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet 2010;86:378–388.
100. Ali Mosrati M, Schrauwen I, Ben Saiid M, Aifa-Hmani M, Fransen E, Mneja M, et al. Genome-wide analysis reveals a novel autosomal-recessive hearing loss locus DFNB80 on chromosome 2p16.1-p21. J Hum Genet 2013;58:98–101.
101. Shahin H, Walsh T, Rayyan AA, Lee MK, Higgins J, Dickel D, et al. Five novel loci for inherited hearing loss mapped by SNP-based homozygosity profiles in Palestinian families. Eur J Hum Genet 2010;18:407–413.
102. Schraders M, Oostrik J, Huygen PL, Strom TM, van Wijk E, Kunst HP, et al. Mutations in PTPRQ are a cause of autosomal-recessive nonsyndromic hearing impairment DFNB84 and associated with vestibular dysfunction. Am J Hum Genet 2010;86:604–610.
103. Yariz KO, Duman D, Zazo Seco C, Dallman J, Huang M, Peters TA, et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am J Hum Genet 2012;91:872–882.
104. Ali RA, Rehman AU, Khan SN, Husnain T, Riazuddin S, Friedman TB, et al. DFNB86, a novel autosomal recessive non-syndromic deafness locus on chromosome 16p13.3. Clin Genet 2012;81:498–500.
105. Rehman AU, Santos-Cortez RL, Morell RJ, Drummond MC, Ito T, Lee K, et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet 2014;94:144–152.
106. Jaworek TJ, Richard EM, Ivanova AA, Giese AP, Choo DI, Khan SN, et al. An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS Genet 2013;9:e1003774.
107. Basit S, Lee K, Habib R, Chen L, Umm e K, Santos-Cortez RL, et al. DFNB89, a novel autosomal recessive nonsyndromic hearing impairment locus on chromosome 16q21-q23.2. Hum Genet 2011;129:379–385.
108. Ali G, Lee K, Andrade PB, Basit S, Santos-Cortez RL, Chen L, et al. Novel autosomal recessive nonsyndromic hearing impairment locus DFNB90 maps to 7p22.1-p15.3. Hum Hered 2011;71:106–112.
109. Sirmaci A, Erbek S, Price J, Huang M, Duman D, Cengiz FB, et al. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet 2010;86:797–804.
110. Tabatabaiefar MA, Alasti F, Shariati L, Farrokhi E, Fransen E, Nooridaloii MR, et al. DFNB93, a novel locus for autosomal recessive moderate-to-severe hearing impairment. Clin Genet 2011;79:594–598.
111. Simon M, Richard EM, Wang X, Shahzad M, Huang VH, Qaiser TA, et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet 2015;11:e1005097.
112. Ansar M, Lee K, Naqvi SK, Andrade PB, Basit S, Santos-Cortez RL, et al. A new autosomal recessive nonsyndromic hearing impairment locus DFNB96 on chromosome 1p36.31-p36.13. J Hum Genet 2011;56:866–868.
113. Mujtaba G, Schultz JM, Imtiaz A, Morell RJ, Friedman TB, Naz S. A mutation of MET, encoding hepatocyte growth factor receptor, is associated with human DFNB97 hearing loss. J Med Genet 2015;52:548–552.
114. Delmaghani S, Aghaie A, Michalski N, Bonnet C, Weil D, Petit C. Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Hum Mol Genet 2012;21:3835–3844.
115. Li J, Zhao X, Xin Q, Shan S, Jiang B, Jin Y, et al. Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum Mutat 2015;36:98–105.
116. Yousaf R, Gu C, Ahmed ZM, Khan SN, Friedman TB, Riazuddin S, et al. Mutations in diphosphoinositol-pentakisphosphate kinase PPIP5K2 are associated with hearing loss in human and mouse. PLoS Genet 2018;14:e1007297.
117. Imtiaz A, Kohrman DC, Naz S. A frameshift mutation in GRXCR2 causes recessively inherited hearing loss. Hum Mutat 2014;35:618–624.
118. Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin JP, et al. EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J Rare Dis 2014;9:55.
119. Seco CZ, Oonk AM, Dominguez-Ruiz M, Draaisma JM, Gandia M, Oostrik J, et al. Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur J Hum Genet 2015;23:189–194.
120. Diaz-Horta O, Subasioglu-Uzak A, Grati M, DeSmidt A, Foster J 2nd, Cao L, et al. FAM65B is a membrane-associated protein of hair cell stereocilia required for hearing. Proc Natl Acad Sci U S A 2014;111:9864–9868.
121. Dahmani M, Ammar-Khodja F, Bonnet C, Lefevre GM, Hardelin JP, Ibrahim H, et al. EPS8L2 is a new causal gene for childhood onset autosomal recessive progressive hearing loss. Orphanet J Rare Dis 2015;10:96.
122. Diaz-Horta O, Abad C, Sennaroglu L, Foster J 2nd, DeSmidt A, Bademci G, et al. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc Natl Acad Sci U S A 2016;113:5993–5998.
123. Walsh T, Shahin H, Elkan-Miller T, Lee MK, Thornton AM, Roeb W, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 2010;87:90–94.
124. Diaz-Horta O, Sirmaci A, Doherty D, Nance W, Arnos K, Pandya A, et al.
GPSM2 mutations in Chudley-McCullough syndrome. Am J Med Genet A 2012;158A:2972–2973.
125. Doherty D, Chudley AE, Coghlan G, Ishak GE, Innes AM, Lemire EG, et al.
GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am J Hum Genet 2012;90:1088–1093.
126. Leon PE, Raventos H, Lynch E, Morrow J, King MC. The gene for an inherited form of deafness maps to chromosome 5q31. Proc Natl Acad Sci U S A 1992;89:5181–5184.
127. Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 1997;278:1315–1318.
128. Coucke P, Van Camp G, Djoyodiharjo B, Smith SD, Frants RR, Padberg GW, et al. Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med 1994;331:425–431.
129. Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, et al. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999;96:437–446.
130. Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 1998;20:370–373.
131. Gao X, Yuan YY, Lin QF, Xu JC, Wang WQ, Qiao YH, et al. Mutation of IFNLR1, an interferon lambda receptor 1, is associated with autosomal-dominant non-syndromic hearing loss. J Med Genet 2018;55:298–306.
132. Chaib H, Lina-Granade G, Guilford P, Plauchu H, Levilliers J, Morgon A, et al. A gene responsible for a dominant form of neurosensory non-syndromic deafness maps to the NSRD1 recessive deafness gene interval. Hum Mol Genet 1994;3:2219–2222.
133. Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaib H, Levi-Acobas F, et al. Connexin 26 gene linked to a dominant deafness. Nature 1998;393:319–320.
134. Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat Genet 1999;23:16–18.
135. Chen AH, Ni L, Fukushima K, Marietta J, O’Neill M, Coucke P, et al. Linkage of a gene for dominant non-syndromic deafness to chromosome 19. Hum Mol Genet 1995;4:1073–1076.
136. Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am J Hum Genet 2004;74:770–776.
137. Zheng J, Miller KK, Yang T, Hildebrand MS, Shearer AE, DeLuca AP, et al. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4). Proc Natl Acad Sci U S A 2011;108:4218–4223.
138. van Camp G, Coucke P, Balemans W, van Velzen D, van de Bilt C, van Laer L, et al. Localization of a gene for non-syndromic hearing loss (DFNA5) to chromosome 7p15. Hum Mol Genet 1995;4:2159–2163.
139. Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet 1998;20:194–197.
140. Lesperance MM, Hall JW 3rd, Bess FH, Fukushima K, Jain PK, Ploplis B, et al. A gene for autosomal dominant nonsyndromic hereditary hearing impairment maps to 4p16.3. Hum Mol Genet 1995;4:1967–1972.
141. Van Camp G, Kunst H, Flothmann K, McGuirt W, Wauters J, Marres H, et al. A gene for autosomal dominant hearing impairment (DFNA14) maps to a region on chromosome 4p16.3 that does not overlap the DFNA6 locus. J Med Genet 1999;36:532–536.
142. Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001;10:2501–2508.
143. Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001;10:2509–2514.
144. Fagerheim T, Nilssen O, Raeymaekers P, Brox V, Moum T, Elverland HH, et al. Identification of a new locus for autosomal dominant non-syndromic hearing impairment (DFNA7) in a large Norwegian family. Hum Mol Genet 1996;5:1187–1191.
145. Wesdorp M, de Koning Gans PA, Schraders M, Oostrik J, Huynen MA, Venselaar H, et al. Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction. Hum Genet 2018;May. 12. [Epub].
https://doi.org/10.1007/s00439-018-1880-5
.
146. Manolis EN, Yandavi N, Nadol JB Jr, Eavey RD, McKenna M, Rosenbaum S, et al. A gene for non-syndromic autosomal dominant progressive postlingual sensorineural hearing loss maps to chromosome 14q12-13. Hum Mol Genet 1996;5:1047–1050.
147. Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 1998;20:299–303.
148. O’Neill ME, Marietta J, Nishimura D, Wayne S, Van Camp G, Van Laer L, et al. A gene for autosomal dominant late-onset progressive non-syndromic hearing loss, DFNA10, maps to chromosome 6. Hum Mol Genet 1996;5:853–856.
149. Wayne S, Robertson NG, DeClau F, Chen N, Verhoeven K, Prasad S, et al. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus. Hum Mol Genet 2001;10:195–200.
150. Tamagawa Y, Kitamura K, Ishida T, Ishikawa K, Tanaka H, Tsuji S, et al. A gene for a dominant form of non-syndromic sensorineural deafness (DFNA11) maps within the region containing the DFNB2 recessive deafness gene. Hum Mol Genet 1996;5:849–852.
151. Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, et al. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet 1997;17:268–269.
152. Brown MR, Tomek MS, Van Laer L, Smith S, Kenyon JB, Van Camp G, et al. A novel locus for autosomal dominant nonsyndromic hearing loss, DFNA13, maps to chromosome 6p. Am J Hum Genet 1997;61:924–927.
153. McGuirt WT, Prasad SD, Griffith AJ, Kunst HP, Green GE, Shpargel KB, et al. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat Genet 1999;23:413–419.
154. Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 1998;279:1950–1954.
155. Fukushima K, Kasai N, Ueki Y, Nishizaki K, Sugata K, Hirakawa S, et al. A gene for fluctuating, progressive autosomal dominant nonsyndromic hearing loss, DFNA16, maps to chromosome 2q23-24.3. Am J Hum Genet 1999;65:141–150.
156. Lalwani AK, Luxford WM, Mhatre AN, Attaie A, Wilcox ER, Castelein CM. A new locus for nonsyndromic hereditary hearing impairment, DFNA17, maps to chromosome 22 and represents a gene for cochleosaccular degeneration. Am J Hum Genet 1999;64:318–323.
157. Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9
. Am J Hum Genet 2000;67:1121–1128.
158. Bonsch D, Scheer P, Neumann C, Lang-Roth R, Seifert E, Storch P, et al. A novel locus for autosomal dominant, non-syndromic hearing impairment (DFNA18) maps to chromosome 3q22 immediately adjacent to the DM2 locus. Eur J Hum Genet 2001;9:165–170.
159. Green GE, Whitehead S, Van Camp G, Smith RJ. Identification of a new locus-DFNA19-for dominant hearing impairment. In : Molecular Biology of Hearing and Deafness Meeting; 1998 Oct 8; Bathesda, MD.
160. Morell RJ, Friderici KH, Wei S, Elfenbein JL, Friedman TB, Fisher RA. A new locus for late-onset, progressive, hereditary hearing loss DFNA20 maps to 17q25. Genomics 2000;63:1–6.
161. Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 2003;73:1082–1091.
162. van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW, et al. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet 2003;40:879–884.
163. Kunst H, Marres H, Huygen P, van Duijnhoven G, Krebsova A, van der Velde S, et al. Non-syndromic autosomal dominant progressive non-specific mid-frequency sensorineural hearing impairment with childhood to late adolescence onset (DFNA21). Clin Otolaryngol Allied Sci 2000;25:45–54.
164. Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet 2001;69:635–640.
165. Salam AA, Hafner FM, Linder TE, Spillmann T, Schinzel AA, Leal SM. A novel locus (DFNA23) for prelingual autosomal dominant nonsyndromic hearing loss maps to 14q21-q22 in a Swiss German kindred. Am J Hum Genet 2000;66:1984–1988.
166. Mosrati MA, Hammami B, Rebeh IB, Ayadi L, Dhouib L, Ben Mahfoudh K, et al. A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. Eur J Med Genet 2011;54:e484–488.
167. Hafner FM, Salam AA, Linder TE, Balmer D, Baumer A, Schinzel AA, et al. A novel locus (DFNA24) for prelingual nonprogressive autosomal dominant nonsyndromic hearing loss maps to 4q35-qter in a large Swiss German kindred. Am J Hum Genet 2000;66:1437–1442.
168. Greene CC, McMillan PM, Barker SE, Kurnool P, Lomax MI, Burmeister M, et al. DFNA25, a novel locus for dominant nonsyndromic hereditary hearing impairment, maps to 12q21-24. Am J Hum Genet 2001;68:254–260.
169. Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van Rybroek JM, et al. Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 2008;83:278–292.
170. Nakano Y, Kelly MC, Rehman AU, Boger ET, Morell RJ, Kelley MW, et al. Defects in the alternative splicing-dependent regulation of REST cause deafness. Cell 2018;174:536–548.e521.
171. Peters LM, Fridell RA, Boger ET, San Agustin TB, Madeo AC, Griffith AJ, et al. A locus for autosomal dominant progressive non-syndromic hearing loss, DFNA27, is on chromosome 4q12-13.1. Clin Genet 2008;73:367–372.
172. Peters LM, Anderson DW, Griffith AJ, Grundfast KM, San Agustin TB, Madeo AC, et al. Mutation of a transcription factor, TFCP2L3, causes progressive autosomal dominant hearing loss, DFNA28. Hum Mol Genet 2002;11:2877–2885.
173. Mangino M, Flex E, Capon F, Sangiuolo F, Carraro E, Gualandi F, et al. Mapping of a new autosomal dominant nonsyndromic hearing loss locus (DFNA30) to chromosome 15q25-26. Eur J Hum Genet 2001;9:667–671.
174. Snoeckx RL, Kremer H, Ensink RJ, Flothmann K, de Brouwer A, Smith RJ, et al. A novel locus for autosomal dominant non-syndromic hearing loss, DFNA31, maps to chromosome 6p21.3. J Med Genet 2004;41:11–13.
175. Chatterjee A, Jalvi R, Pandey N, Rangasayee R, Anand A. A novel locus DFNA59 for autosomal dominant nonsyndromic hearing loss maps at chromosome 11p14.2-q12.3. Hum Genet 2009;124:669–675.
176. Bonsch D, Schmidt CM, Scheer P, Bohlender J, Neumann C, Am Zehnhoff-Dinnesen A, et al. A new gene locus for an autosomal-dominant non-syndromic hearing impairment (DFNA 33) is situated on chromosome 13q34-qter. HNO 2009;57:371–376.
177. Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al.
NLRP3 mutation and cochlear auto-inflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci U S A 2017;114:E7766–E7775.
178. Booth KT, Askew JW, Talebizadeh Z, Huygen PLM, Eudy J, Kenyon J, et al. Splice-altering variant in
COL11A1 as a cause of nonsyndromic hearing loss DFNA37. Genet Med 2018;Sep. 24. [Epub].
https://doi.org/10.1038/s41436-018-0285-0
.
179. Xia J, Deng H, Feng Y, Zhang H, Pan Q, Dai H, et al. A novel locus for autosomal dominant nonsyndromic hearing loss identified at 5q31.1-32 in a Chinese pedigree. J Hum Genet 2002;47:635–640.
180. Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, et al. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet 2003;72:73–82.
181. Blanton SH, Liang CY, Cai MW, Pandya A, Du LL, Landa B, et al. A novel locus for autosomal dominant non-syndromic deafness (DFNA41) maps to chromosome 12q24-qter. J Med Genet 2002;39:567–570.
182. Yan D, Zhu Y, Walsh T, Xie D, Yuan H, Sirmaci A, et al. Mutation of the ATP-gated P2X(2) receptor leads to progressive hearing loss and increased susceptibility to noise. Proc Natl Acad Sci U S A 2013;110:2228–2233.
183. Flex E, Mangino M, Mazzoli M, Martini A, Migliosi V, Colosimo A, et al. Mapping of a new autosomal dominant non-syndromic hearing loss locus (DFNA43) to chromosome 2p12. J Med Genet 2003;40:278–281.
184. Modamio-Hoybjor S, Moreno-Pelayo MA, Mencia A, del Castillo I, Chardenoux S, Armenta D, et al. A novel locus for autosomal dominant nonsyndromic hearing loss (DFNA44) maps to chromosome 3q28-29. Hum Genet 2003;112:24–28.
185. Modamio-Hoybjor S, Mencia A, Goodyear R, del Castillo I, Richardson G, Moreno F, et al. A mutation in CCDC50, a gene encoding an effector of epidermal growth factor-mediated cell signaling, causes progressive hearing loss. Am J Hum Genet 2007;80:1076–1089.
186. D’Adamo P, Donaudy F, D’Eustacchio A, Di Iorio E, Melchionda S, Gasparini P. A new locus (DFNA47) for autosomal dominant non-syndromic inherited hearing loss maps to 9p21-22 in a large Italian family. Eur J Hum Genet 2003;11:121–124.
187. D’Adamo P, Pinna M, Capobianco S, Cesarani A, D’Eustacchio A, Fogu P, et al. A novel autosomal dominant non-syndromic deafness locus (DFNA48) maps to 12q13-q14 in a large Italian family. Hum Genet 2003;112:319–320.
188. Donaudy F, Ferrara A, Esposito L, Hertzano R, Ben-David O, Bell RE, et al. Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am J Hum Genet 2003;72:1571–1577.
189. Moreno-Pelayo MA, Modamio-Hoybjor S, Mencia A, del Castillo I, Chardenoux S, Fernandez-Burriel M, et al. DFNA49, a novel locus for autosomal dominant non-syndromic hearing loss, maps proximal to DFNA7/DFNM1 region on chromosome 1q21-q23. J Med Genet 2003;40:832–836.
190. Modamio-Hoybjor S, Moreno-Pelayo MA, Mencia A, del Castillo I, Chardenoux S, Morais D, et al. A novel locus for autosomal dominant nonsyndromic hearing loss, DFNA50, maps to chromosome 7q32 between the DFNB17 and DFNB13 deafness loci. J Med Genet 2004;41:e14.
191. Mencia A, Modamio-Hoybjor S, Redshaw N, Morin M, Mayo-Merino F, Olavarrieta L, et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 2009;41:609–613.
192. Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, et al. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet 2010;87:101–109.
193. Yan D, Ke X, Blanton SH, Ouyang XM, Pandya A, Du LL, et al. A novel locus for autosomal dominant non-syndromic deafness, DFNA53, maps to chromosome 14q11.2-q12. J Med Genet 2006;43:170–174.
194. Gurtler N, Kim Y, Mhatre A, Schlegel C, Mathis A, Lalwani AK. DFNA54, a third locus for low-frequency hearing loss. J Mol Med (Berl) 2004;82:775–780.
195. Zhao Y, Zhao F, Zong L, Zhang P, Guan L, Zhang J, et al. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss. PLoS One 2013;8:e69549.
196. Bonsch D, Schmidt CM, Scheer P, Bohlender J, Neumann C, am Zehnhoff-Dinnesen A, et al. A new locus for an autosomal dominant, non-syndromic hearing impairment (DFNA57) located on chromosome 19p13.2 and overlapping with DFNB15. HNO 2008;56:177–182.
197. Lezirovitz K, Braga MC, Thiele-Aguiar RS, Auricchio MT, Pearson PL, Otto PA, et al. A novel autosomal dominant deafness locus (DFNA58) maps to 2p12-p21. Clin Genet 2009;75:490–493.
198. Ouyang XM, Yan D, Du LL. A novel locus for autosomal dominant non-syndromic hearing loss maps to chromosome 2q213-q241. In : Midwinter Meeting for the Association for Research in Otolaryngology; 2007 Feb 11–15; Denver, CO.
199. Cheng J, Zhu Y, He S, Lu Y, Chen J, Han B, et al. Functional mutation of SMAC/DIABLO, encoding a mitochondrial proapoptotic protein, causes human progressive hearing loss DFNA64. Am J Hum Genet 2011;89:56–66.
200. Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, et al.
TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum Mutat 2014;35:819–823.
201. Nyegaard M, Rendtorff ND, Nielsen MS, Corydon TJ, Demontis D, Starnawska A, et al. A novel locus harbouring a functional CD164 nonsense mutation identified in a large Danish family with nonsyndromic hearing impairment. PLoS Genet 2015;11:e1005386.
202. Thoenes M, Zimmermann U, Ebermann I, Ptok M, Lewis MA, Thiele H, et al.
OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J Rare Dis 2015;10:15.
203. Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PL, et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet 2015;11:e1005137.
204. Zazo Seco C, Serrao de Castro L, van Nierop JW, Morin M, Jhangiani S, Verver EJ, et al. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am J Hum Genet 2015;97:647–660.
205. Gao J, Wang Q, Dong C, Chen S, Qi Y, Liu Y. Whole exome sequencing identified MCM2 as a novel causative gene for autosomal dominant nonsyndromic deafness in a Chinese family. PLoS One 2015;10:e0133522.
206. Eisenberger T, Di Donato N, Decker C, Delle Vedove A, Neuhaus C, Nurnberg G, et al. A C-terminal nonsense mutation links PTPRQ with autosomal-dominant hearing loss, DFNA73. Genet Med 2018;20:614–621.
207. Eisenberger T, Di Donato N, Baig SM, Neuhaus C, Beyer A, Decker E, et al. Targeted and genomewide NGS data disqualify mutations in MYO1A, the “DFNA48 gene”, as a cause of deafness. Hum Mutat 2014;35:565–570.
208. Liu X, Han D, Li J, Han B, Ouyang X, Cheng J, et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am J Hum Genet 2010;86:65–71.
209. de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4
. Science 1995;267:685–688.
210. Schraders M, Haas SA, Weegerink NJ, Oostrik J, Hu H, Hoefsloot LH, et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am J Hum Genet 2011;88:628–634.
211. Huebner AK, Gandia M, Frommolt P, Maak A, Wicklein EM, Thiele H, et al. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am J Hum Genet 2011;88:621–627.
212. del Castillo I, Villamar M, Sarduy M, Romero L, Herraiz C, Hernandez FJ, et al. A novel locus for non-syndromic sensorineural deafness (DFN6) maps to chromosome Xp22. Hum Mol Genet 1996;5:1383–1387.
213. Zong L, Guan J, Ealy M, Zhang Q, Wang D, Wang H, et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J Med Genet 2015;52:523–531.
214. Rost S, Bach E, Neuner C, Nanda I, Dysek S, Bittner RE, et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6
. Eur J Hum Genet 2014;22:208–215.
215. Wang QJ, Lu CY, Li N, Rao SQ, Shi YB, Han DY, et al. Y-linked inheritance of non-syndromic hearing impairment in a large Chinese family. J Med Genet 2004;41:e80.
216. Bykhovskaya Y, Estivill X, Taylor K, Hang T, Hamon M, Casano RA, et al. Candidate locus for a nuclear modifier gene for maternally inherited deafness. Am J Hum Genet 2000;66:1905–1910.
217. Schoen CJ, Emery SB, Thorne MC, Ammana HR, Sliwerska E, Arnett J, et al. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila. Proc Natl Acad Sci U S A 2010;107:13396–13401.
218. Kim TB, Isaacson B, Sivakumaran TA, Starr A, Keats BJ, Lesperance MM. A gene responsible for autosomal dominant auditory neuropathy (AUNA1) maps to 13q14-21. J Med Genet 2004;41:872–876.
219. Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016;135:441–450.
220. Hilgert N, Smith RJ, Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr Mol Med 2009;9:546–564.
221. Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC, Pirson Y, et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994;8:77–81.
222. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990;248:1224–1227.
223. Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 1997;15:157–164.
224. Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 2007;80:800–804.
225. Ruf RG, Berkman J, Wolf MT, Nurnberg P, Gattas M, Ruf EM, et al. A gene locus for branchio-otic syndrome maps to chromosome 14q21.3-q24.3. J Med Genet 2003;40:515–519.
226. Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 2004;41:e94.
227. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004;36:955–957.
228. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 1997;15:186–189.
229. Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JF, Bathen J, et al. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 1997;6:2179–2185.
230. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, et al.
KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet 1997;17:267–268.
231. Berger W, van de Pol D, Warburg M, Gal A, Bleeker-Wagemakers L, de Silva H, et al. Mutations in the candidate gene for Norrie disease. Hum Mol Genet 1992;1:461–465.
232. Chen ZY, Hendriks RW, Jobling MA, Powell JF, Breakefield XO, Sims KB, et al. Isolation and characterization of a candidate gene for Norrie disease. Nat Genet 1992;1:204–208.
233. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 1997;17:411–422.
234. Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet 2007;80:1055–1063.
235. Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, et al. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet 2009;84:651–657.
236. Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet 2010;87:282–288.
237. Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet 2013;92:605–613.
238. Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 2013;92:614–620.
239. Morino H, Pierce SB, Matsuda Y, Walsh T, Ohsawa R, Newby M, et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014;83:2054–2061.
240. Chatzispyrou IA, Alders M, Guerrero-Castillo S, Zapata Perez R, Haagmans MA, Mouchiroud L, et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet 2017;26:2541–2550.
241. Ahmad NN, Ala-Kokko L, Knowlton RG, Jimenez SA, Weaver EJ, Maguire JI, et al. Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci U S A 1991;88:6624–6627.
242. Richards AJ, Yates JR, Williams R, Payne SJ, Pope FM, Scott JD, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet 1996;5:1339–1343.
243. Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, et al. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995;80:431–437.
244. Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, Fukuoka H, Wagatsuma M, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet 2006;79:449–457.
245. Baker S, Booth C, Fillman C, Shapiro M, Blair MP, Hyland JC, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A 2011;155A:1668–1672.
246. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. The Treacher Collins Syndrome Collaborative Group. Nat Genet 1996;12:130–136.
247. Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet 2011;43:20–22.
248. Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995;374:60–61.
249. Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet 2000;26:51–55.
250. Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet 2001;27:108–112.
251. Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, et al. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet 2001;69:25–34.
252. Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet 2003;12:463–471.
253. Ahmed ZM, Riazuddin S, Khan SN, Friedman PL, Riazuddin S, Friedman TB. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin Genet 2009;75:86–91.
254. Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998;280:1753–1757.
255. Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 2004;74:357–366.
256. Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 2007;121:203–211.
257. Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 2001;69:673–684.
258. Bondurand N, Kuhlbrodt K, Pingault V, Enderich J, Sajus M, Tommerup N, et al. A molecular analysis of the yemenite deaf-blind hypopigmentation syndrome: SOX10 dysfunction causes different neurocristopathies. Hum Mol Genet 1999;8:1785–1789.
259. Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet 1994;8:251–255.
260. Sanchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet 2002;11:3231–3236.
261. Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, et al. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet 2007;81:1169–1185.
262. Zlotogora J, Lerer I, Bar-David S, Ergaz Z, Abeliovich D. Homozygosity for Waardenburg syndrome. Am J Hum Genet 1995;56:1173–1178.
263. Attie T, Till M, Pelet A, Amiel J, Edery P, Boutrand L, et al. Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum Mol Genet 1995;4:2407–2409.
264. Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet 1996;12:442–444.
265. Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE, Prehu MO, Puliti A, et al. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat Genet 1998;18:171–173.
266. Robertson NG, Khetarpal U, Gutierrez-Espeleta GA, Bieber FR, Morton CC. Isolation of novel and known genes from a human fetal cochlear cDNA library using subtractive hybridization and differential screening. Genomics 1994;23:42–50.
267. Sakuma N, Moteki H, Takahashi M, Nishio SY, Arai Y, Yamashita Y, et al. An effective screening strategy for deafness in combination with a next-generation sequencing platform: a consecutive analysis. J Hum Genet 2016;61:253–261.
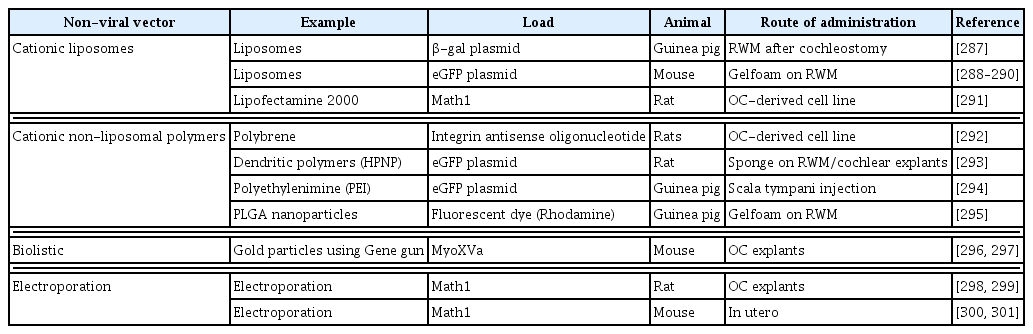
268. Liu H, Hao J, Li KS. Current strategies for drug delivery to the inner ear. Acta Pharm Sin B 2013;3:86–96.
269. Lee MY, Kurioka T, Nelson MM, Prieskorn DM, Swiderski DL, Takada Y, et al. Viral-mediated Ntf3 overexpression disrupts innervation and hearing in nondeafened guinea pig cochleae. Mol Ther Methods Clin Dev 2016;3:16052.
270. Takada Y, Takada T, Lee MY, Swiderski DL, Kabara LL, Dolan DF, et al. Ototoxicity-induced loss of hearing and inner hair cells is attenuated by HSP70 gene transfer. Mol Ther Methods Clin Dev 2015;2:15019.
271. Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron 2012;75:283–293.
272. Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, et al. Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol Ther 2016;24:17–25.
273. Geschwind MD, Hartnick CJ, Liu W, Amat J, Van De Water TR, Federoff HJ. Defective HSV-1 vector expressing BDNF in auditory ganglia elicits neurite outgrowth: model for treatment of neuron loss following cochlear degeneration. Hum Gene Ther 1996;7:173–182.
274. Izumikawa M, Minoda R, Kawamoto K, Abrashkin KA, Swiderski DL, Dolan DF, et al. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med 2005;11:271–276.
275. Kawamoto K, Ishimoto S, Minoda R, Brough DE, Raphael Y. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J Neurosci 2003;23:4395–4400.
276. Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol 2017;35:264–272.
277. Pietola L, Aarnisalo AA, Joensuu J, Pellinen R, Wahlfors J, Jero J. HOX-GFP and WOX-GFP lentivirus vectors for inner ear gene transfer. Acta Otolaryngol 2008;128:613–620.
278. Schlecker C, Praetorius M, Brough DE, Presler RG Jr, Hsu C, Plinkert PK, et al. Selective atonal gene delivery improves balance function in a mouse model of vestibular disease. Gene Ther 2011;18:884–890.
279. Shibata SB, Di Pasquale G, Cortez SR, Chiorini JA, Raphael Y. Gene transfer using bovine adeno-associated virus in the guinea pig cochlea. Gene Ther 2009;16:990–997.
280. Staecker H, Liu W, Malgrange B, Lefebvre PP, Van De Water TR. Vector-mediated delivery of bcl-2 prevents degeneration of auditory hair cells and neurons after injury. ORL J Otorhinolaryngol Relat Spec 2007;69:43–50.
281. Raphael Y, Frisancho JC, Roessler BJ. Adenoviral-mediated gene transfer into guinea pig cochlear cells in vivo. Neurosci Lett 1996;207:137–141.
282. Yagi M, Magal E, Sheng Z, Ang KA, Raphael Y. Hair cell protection from aminoglycoside ototoxicity by adenovirus-mediated overexpression of glial cell line-derived neurotrophic factor. Hum Gene Ther 1999;10:813–823.
283. Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol 2017;35:280–284.
284. Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, et al. Tmc gene therapy restores auditory function in deaf mice. Sci Transl Med 2015;7:295ra108.
285. Di Pasquale G, Rzadzinska A, Schneider ME, Bossis I, Chiorini JA, Kachar B. A novel bovine virus efficiently transduces inner ear neuroepithelial cells. Mol Ther 2005;11:849–855.
286. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 2003;4:346–358.
287. Wareing M, Mhatre AN, Pettis R, Han JJ, Haut T, Pfister MH, et al. Cationic liposome mediated transgene expression in the guinea pig cochlea. Hear Res 1999;128:61–69.
288. Jero J, Mhatre AN, Tseng CJ, Stern RE, Coling DE, Goldstein JA, et al. Cochlear gene delivery through an intact round window membrane in mouse. Hum Gene Ther 2001;12:539–548.
289. Jero J, Tseng CJ, Mhatre AN, Lalwani AK. A surgical approach appropriate for targeted cochlear gene therapy in the mouse. Hear Res 2001;151:106–114.
290. Staecker H, Li D, O’Malley BW Jr, Van De Water TR. Gene expression in the mammalian cochlea: a study of multiple vector systems. Acta Otolaryngol 2001;121:157–163.
291. Zhang W, Zhang Y, Sood R, Ranjan S, Surovtseva E, Ahmad A, et al. Visualization of intracellular trafficking of Math1 protein in different cell types with a newly-constructed non-viral gene delivery plasmid. J Gene Med 2011;13:134–144.
292. Toyama K, Ozeki M, Hamajima Y, Lin J. Expression of the integrin genes in the developing cochlea of rats. Hear Res 2005;201:21–26.
293. Zhang W, Zhang Y, Lobler M, Schmitz KP, Ahmad A, Pyykko I, et al. Nuclear entry of hyperbranched polylysine nanoparticles into cochlear cells. Int J Nanomedicine 2011;6:535–546.
294. Tan BT, Foong KH, Lee MM, Ruan R. Polyethylenimine-mediated cochlear gene transfer in guinea pigs. Arch Otolaryngol Head Neck Surg 2008;134:884–891.
295. Tamura T, Kita T, Nakagawa T, Endo T, Kim TS, Ishihara T, et al. Drug delivery to the cochlea using PLGA nanoparticles. Laryngoscope 2005;115:2000–2005.
296. Belyantseva IA, Boger ET, Friedman TB. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A 2003;100:13958–13963.
297. Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, et al. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 2005;7:148–156.
298. Woods C, Montcouquiol M, Kelley MW. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci 2004;7:1310–1318.
299. Zheng JL, Gao WQ. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci 2000;3:580–586.
300. Brigande JV, Gubbels SP, Woessner DW, Jungwirth JJ, Bresee CS. Electroporation-mediated gene transfer to the developing mouse inner ear. Methods Mol Biol 2009;493:125–139.
301. Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV. Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 2008;455:537–541.
302. Wrobel I, Collins D. Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochim Biophys Acta 1995;1235:296–304.
303. Gao X, Kim KS, Liu D. Nonviral gene delivery: what we know and what is next. AAPS J 2007;9:E92–E104.
304. Belyantseva IA. Helios Gene Gun-mediated transfection of the inner ear sensory epithelium. Methods Mol Biol 2009;493:103–123.
305. Heller LC, Ugen K, Heller R. Electroporation for targeted gene transfer. Expert Opin Drug Deliv 2005;2:255–268.
306. Ahmed H, Shubina-Oleinik O, Holt JR. Emerging gene therapies for genetic hearing loss. J Assoc Res Otolaryngol 2017;18:649–670.
307. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494–498.
308. Hill K, Yuan H, Wang X, Sha SH. Noise-induced loss of hair cells and cochlear synaptopathy are mediated by the activation of AMPK
. J Neurosci 2016;36:7497–7510.
309. Shibata SB, Ranum PT, Moteki H, Pan B, Goodwin AT, Goodman SS, et al. RNA interference prevents autosomal-dominant hearing loss. Am J Hum Genet 2016;98:1101–1113.
310. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823.
311. Lee MY, Park YH. Potential of gene and cell therapy for inner ear hair cells. Biomed Res Int 2018;20188137614.
312. Zou B, Mittal R, Grati M, Lu Z, Shu Y, Tao Y, et al. The application of genome editing in studying hearing loss. Hear Res 2015;327:102–108.
313. Hirano S, Nishimasu H, Ishitani R, Nureki O. Structural basis for the altered PAM specificities of engineered CRISPR-Cas9. Mol Cell 2016;61:886–894.
314. Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. Gene therapy clinical trials worldwide to 2017: An update
. J Gene Med 2018;20:e3015.
315. Lopes VS, Williams DS. Gene therapy for the retinal degeneration of Usher syndrome caused by mutations in MYO7A. Cold Spring Harb Perspect Med 2015;5:a017319.